 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yohan Santin | + 2726 word(s) | 2726 | 2020-11-24 02:56:07 | | | |
| 2 | Yohan Santin | -13 word(s) | 2713 | 2020-11-24 14:52:39 | | | | |
| 3 | Camila Xu | Meta information modification | 2713 | 2020-11-25 04:50:23 | | |
Video Upload Options
Cellular senescence is a state of cell cycle arrest induced by repetitive cell mitoses or different stresses, which is implicated in various physiological or pathological processes. The beneficial or adverse effects of senescent cells depend on their transitory or persistent state. Recently, an increase in senescent cell burden has been reported in renal disorders. Here, we will summarize the molecular mechanisms of senescence and their implication in renal diseases. We will also discuss the differential impacts of transient versus persistent status of cellular senescence.
1. Overview of Cellular Senescence
Cellular senescence refers to a state of stable cell cycle arrest that can be initiated by various stresses despite the presence of growth-promoting stimuli. Senescence can occur in multiple contexts across tissue and organ lifespans. Within this line, acute senescence generated early in life provides physiologically-appropriate responses in the developing embryo during organogenesis. This transient phenotype also plays key roles in tissue homeostasis, wound healing, and regeneration. In addition, senescence-related growth arrest prevents tumorigenesis and neoplastic transformation [1][2]. Conversely, chronic accumulation of senescent cells is increasingly recognized as a driver of various features of aging, including age-related diseases and tissue deterioration [3].
Major triggers of senescence include repeated cell division and telomere shortening, first described by Leonard Hayflick in 1961 (now referred to as replicative senescence), but also stressors such as oncogenic mutations, metabolic and oxidative stresses, mitochondrial dysfunction, and inflammation (referred to as stress-induced premature senescence or SIPS) [4]. Senescent cells are often characterized by a persistent DNA damage response (DDR) and the triggering of cell signaling cascades involved in DNA repair and cell cycle arrest, including chronic ATM (ataxia-telangiectasia-mutated) or ATR (ataxia-telangiectasia- and Rad3-related) kinase activation. A typical example of the consequences of DNA damage is the generation of the phosphorylated form of the H2A histone family member X (γH2AX) by ATM. These pathways converge to cell cycle arrest and senescence, through activation of p53/p21CIP1 and p16INK4A that inhibit cyclin-dependent kinases (CDKs) and retinoblastoma protein (RB), enhancing checkpoint activity and inducing G1/S (and occasionally G2/M) cell cycle arrest [5] (Figure 1).
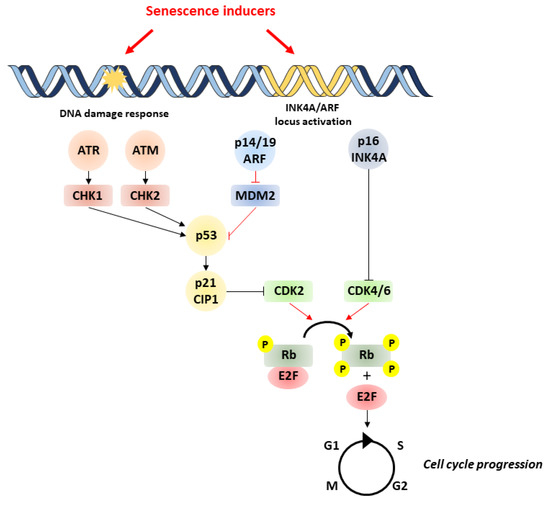
Figure 1. Pathways involved in cell senescence. Senescence is induced by various stressors, which trigger DNA damage and subsequent activation of p53/p21 and p16/pRb pathways. p53 activation is achieved by phosphorylation by ATM/ATR and checkpoint kinases Chk1/Chk2. Likewise, p53 activity can be increased by binding of p14/P19ARF product of the INK4a locus to MDM2 preventing degradation of p53. p53 can induce senescence by activating p21, which inhibits CDK2, leading to the hypophosphorylation of Rb. In addition to p53, the accumulation of the tumor-suppressor p16INK4A also leads to cell cycle arrest through the inhibition of CDK4/CDK6 and subsequent hypophosphorylation of Rb. This enables Rb to bind to E2F, inhibiting cell cycle progression. ATM, ataxia-telangiectasia-mutated kinase; ATR, ataxia-telangiectasia- and ATM-Rad3-related kinase; MDM2, murine double minute 2; Rb, retinoblastoma.
Despite the cell cycle arrest, senescent cells remain metabolically active, releasing a particular secretome that can affect neighboring cells and ultimately tissue function. This phenotype is defined as “senescence-associated secretory phenotype” (SASP) and relies on the production of a specific “senescence-messaging secretome” (SMS) including pro-fibrotic and pro-inflammatory factors like IL-1β, IL-6, TGFβ, PAI-1, or CCN2, which can act in a paracrine and autocrine fashion [6][7]. Although pro-inflammatory cytokines and chemokines are relatively conserved SMS components, SMS composition can vary depending on the biological context, often reflecting both the origin of the senescent cells and the initiating stimuli. Therefore, the functions attributed to the SMS are very diverse and depend on the transient versus persistent status of senescent cells, the nature of the SMS, and the surrounding environment of the cells subjected to the senescent secretome. For example, by an autocrine mechanism, the SMS reinforces the senescence-associated growth arrest by implementing a positive-feedback loop, leading to persistence and propagation of senescence within tissues [8]. On the other hand, the pro-inflammatory nature and inflammatory mediators of the SMS are powerful drivers of tumor progression through a paracrine mechanism.
Other key features of senescent cells, including morphological and metabolic changes, have been described. Indeed, in addition to enlarged size, flattened shape, and sometimes multiple or enlarged nuclei, senescent cells exhibit striking chromatin changes, through the formation of senescence-associated heterochromatic foci (SAHFs). These senescence-specific heterochromatic compartments are enriched in chromatin modifications and sequester genes implicated in cell-cycle control, reinforcing the senescence-associated growth arrest. Of note, genomic regions contained in the SAHFs are found in lamina-associated domains (LADs) [9]. Upon induction of senescence, lamin B1 is downregulated, making these LADs detach from the nuclear periphery and cluster within the nuclei, participating in the disruption of nuclear envelope integrity. In addition to nuclear modifications, senescent cells usually display extensive vacuolization due to an enhanced lysosomal content, reflected by increased levels of β-galactosidase activity. This enzymatic activity is found in non-senescent cells under physiological conditions (pH 4.0–4.5), but it is significantly increased in senescent cells and detectable at pH 6.0 (suboptimal conditions). Importantly, senescence-associated β-galactosidase (SA-β-gal) activity can be measured both in vitro and in vivo, and is considered a key hallmark of senescence [10]. Of note, lipofuscin, an aggregate of oxidized proteins, lipids, and metals that are known to accumulate in aged tissues, was recently reported to co-localize with SA-β-gal activity in senescent cells, and could represent another easily measurable marker of senescent cells [11].
Metabolic changes displayed by senescent cells include increases in glycolysis, mitochondrial metabolism, and autophagy dysfunction, while their roles remain controversial. For instance, the production of SASP components has been shown to rely on enhanced ATP production mediated by increased mitochondrial metabolism and glycolysis, but finally leading to a massive proteotoxic stress [12]. Interestingly, the latter can be attenuated by activating autophagy. On the other hand, some studies have shown that inhibition of autophagy facilitates senescence [13]. Within this line, mitochondrial autophagy (mitophagy) is usually decreased in senescent cells, resulting in an altered mitochondrial network, accumulation of dysfunctional mitochondria, and ROS-induced senescence, contributing to aging-related metabolic dysfunction [14][15].
An increase in senescent cell burden has been associated with age-related disorders, chronic diseases, and accelerated mortality [16]. Within this line, cellular senescence has been linked to obesity-related metabolic derangements [17], chronic lung diseases [18], cardiovascular aging [19], as well as neurodegenerative disorders like Alzheimer’s [20] and Parkinson’s [21]. Recently, senescence has drawn a particular attention in the field of urogenital disorders, which affect the urinary tract or reproductive organs, and have a higher incidence in the elderly population. Indeed, studies reported an increase in senescent cell burden in renal diseases and in aging-associated lower urinary tract symptoms [22][23][24][25][26]. However, the role of cellular senescence in such disorders is still unclear.
2. Senescence in the Kidneys
Renal aging is characterized by a loss in renal mass (cortical glomeruli and tubules) and impaired renal function, as well as various histological changes including glomerulosclerosis, tubulointerstitial fibrosis, and nephrosclerosis [27]. These phenotypic changes are associated with a decline of kidney function and diminished proliferative response of tubular cells (and to a lesser extent of glomerular and interstitial cells), which correlate with markers of cellular senescence such as p16INK4A, SA-β-gal, and telomere shortening [22][23]. Age-associated changes of the kidney are important not only because normal aging alters renal function, but also because of the high frequency of renal diseases in the elderly population. In particular, chronic kidney disease (CKD), defined by the persistent loss of kidney function, currently affects about 13.4% of the global population [28]. Importantly, CKD is regarded as an accelerated aging of the kidney and an independent risk factor for cardiovascular events in the elderly, often leading to end-stage renal disease (ESRD) [29]. Of note, similar pathogenic mechanisms are involved in both aging and CKD onset, such as an increase in senescent cell burden as well as the secretion of pro-fibrotic and pro-inflammatory factors. This latter feature, known in chronic renal failure as CKD-associated secretory phenotype (CASP), shares many similarities with SASP regarding the secreted factors, which may be some mediators of the crosstalk between cellular senescence and CKD [30][31].
2.1. Acute Renal Injuries
Acute damage of tubular epithelial cells like ischemia-reperfusion injury (IRI) or acute kidney injury (AKI) are primary drivers of renal injury and CKD, especially in elderly patients who have an increased propensity to develop progressive CKD [32]. Notch-induced senescent state in proximal tubules triggers maladaptive repair and leads to lower outcome of old kidneys after IRI [33]. Within this line, mouse models of telomerase deficiency, with higher telomere shortening and accelerated senescence, exhibited delayed renal tubular regeneration and increased apoptosis after IRI [34]. Furthermore, p16INK4A knockout mice show increased epithelial proliferation and functional recovery after IRI, meaning that post-injury renal recovery is improved by lowering the number of senescent cells and improved regenerative capacity of aged kidneys [35]. This idea is further supported in AKI, where kidneys with a high content of senescent cells are more prone to develop CKD due to incomplete recovery and development of tubulointerstitial fibrosis [36]. Nevertheless, some conflicting results highlighted the more intricate role of cell senescence in kidney injury. This is well illustrated by the higher susceptibility of p21 knockout mice to ischemia-induced acute renal failure [37]. Similar results were observed in IRI, where senescence induction by CDK4/CDK6 inhibition ameliorated kidney function following injury [38].
Overall, these observations support a dual role of senescent cells in response to acute renal injuries. One could hypothesize that activation of senescence during an acute stress would limit damage propagation by transiently decreasing cell cycle activity, thereby lessening kidney impairment and mortality. However, if senescent cells accumulate after the stress, it is likely that they negatively influence local milieu and further deteriorate organ function. In any case, further studies are needed to better characterize the roles of senescent cells at various time points and in different models of acute renal injuries.
2.2. Chronic Renal Diseases
ESRD is the most common and serious consequence of diabetes, a pathological condition where damage to the kidneys usually occurs over many years. Importantly, diabetic nephropathy and chronic renal disease are characterized by a progressive onset of fibrosis together with a persistent and low-grade inflammation, both of them being hallmark features of senescent cells and related SASP. Of note, kidneys with type 2 diabetic nephropathy displayed an accelerated senescent phenotype in tubule cells and, to a lesser extent, podocytes, as indicated by increased p16 expression and SA-β-gal activity [39]. A similar senescent phenotype was observed when proximal tubule cell cultures were incubated under high-glucose media [39]. Accordingly, streptozotocin-induced hyperglycemia increased senescent cell burden in mouse kidneys in the early stage of type 1 diabetes, characterized by an increase in the renal expression of p21 and SA-β-gal staining in tubular epithelial cells, as well as the acquisition of SASP in endothelial cells and macrophages [40][41]. Interestingly, SASP mediators were also elevated in serum of patients in early stage diabetic nephropathy [42]. An impaired immune system (e.g., due to aging) may cause senescent cells to escape immune clearance and further maintain a senescent secretome [43]. SMS components could therefore constitute a major source of inflammatory factors in diabetes, likely contributing to fueling the low-grade inflammation commonly observed in this pathology. Importantly, the clearance of senescent cells in a mouse model of obesity-induced metabolic dysfunction improved renal podocyte function, reduced microalbuminuria, and lowered circulating inflammatory mediators [17]. Overall, these results suggest that hyperglycemia is a key driver of senescence, the latter being a potential contributor to the development of diabetic nephropathy and consequent ESRD.
A major feature in ESRD is kidney fibrosis, which develops from a variety of diseases and leads to loss of renal function. Senescence is associated with renal disease progression, and accelerated tubular cell senescence leads to maladaptive repair and contributes to the pathogenesis of renal fibrosis [36]. Interesting data indicated that aged mice 6 weeks post-reperfusion had extensive tubulo-interstitial fibrosis and leukocyte infiltration, which correlated with increased p53 expression and SA-β-gal positive cells in kidney tubules [44]. In addition, AKI-induced senescence of proximal tubular cells produce pro-fibrotic growth factors that are capable of stimulating fibroblast proliferation and collagen production [45]. In human proximal tubular epithelial cell line and primary renal tubular cells, Wnt9a upregulated the levels of p16INK4A, p19ARF, p53, and p21, and induced TGF-β1 production by senescent tubular cells, finally promoting proliferation and activation of kidney fibroblasts. Notably, Wnt9a-induced renal fibrosis was inhibited by silencing of p16INK4A in an IRI mouse model [46]. These results strengthen the general concept that senescent cells that appear after renal injury contribute to a pro-fibrotic and pro-inflammatory milieu through their SASP components, further promoting the gradual accumulation of renal fibrosis [47]. Nevertheless, contrary to the well-described detrimental effect of cellular senescence, there is evidence regarding a beneficial role of senescent cells in renal fibrosis. Within this line, Wolstein and coworkers have shown that p16INK4A knockout mice spontaneously exhibited mesangial cell proliferation, myofibroblast differentiation, and increased matrix deposition in kidneys [48]. In addition, after unilateral ureteral obstruction, p16INK4A knockout mice had enhanced tubular and interstitial cell proliferation, lower collecting duct apoptosis, greater collagen and fibronectin deposition, and no SA-β-gal staining compared with WT mice. These results suggest that p16INK4A regulates cell proliferation and limits matrix production, thereby mitigating post-injury fibrosis [48]. Altogether, these discrepancies suggest a dual role of cellular senescence in the onset of renal fibrosis, meaning that further studies will be needed to elucidate the mechanisms by which senescent tubular cells promote or prevent matrix deposition.
As stated before, CKD can lead to ESRD if the renal function gradually deteriorates. In this case, alternative therapies such as kidney transplant are necessary for patients with ESRD. Studies on humans have correlated the levels of pre-transplant senescent cells in the kidneys with subsequent chronic allograft nephropathy [49][50]. Furthermore, in experimental rat models of transplant rejection, acute rejection was characterized by telomere shortening and consequent p21 and p16 overexpression, while SA-β-gal staining was found in tubular epithelial cells of allografts with chronic rejection [51]. Conversely, p16INK4A deletion in mice led to improved renal function and resulted in superior recipient survival in a life-supporting transplant model [52]. Data in humans showed that telomere length in biopsies collected in the peri-transplant period predicted the long-term kidney allograft function, and telomere shortening was linked to complications of kidney transplantation including delayed graft function, acute rejection, and chronic allograft dysfunction [53]. Of note, senescence may also explain some of the impacts of age on graft outcome. Indeed, kidney transplants from aged donors exhibit a higher number of senescent cells and have a lower graft survival compared to “young kidneys” [54]. In addition, increased numbers of senescent cells in an older graft reduce the regenerative capacity of the transplanted kidney in response to injury [55]. Altogether, these results support a crucial role of senescence in kidney transplant, and the level of senescence before transplantation has been shown to be a good predictor of postoperative kidney function, suggesting that biological age is an important prognostic determinant for renal transplant outcome [56].
2.3. Cancers
Senescence induction by oncogene activation has long been recognized as a potent tumor-suppressive mechanism, thereby preventing the expansion of damaged and pre-neoplastic cells [57]. In line with this, deletion of the Apc tumor suppressor gene induces kidney senescence which inhibits the formation of renal carcinomas [58]. Interestingly, in addition to Apc deletion, loss of p21 or p16INK4A was required to initiate renal carcinoma, supporting the anti-neoplastic role of senescence [58]. Besides, a recent study showed that calcitriol-induced histone demethylase JMJD3 increased p16INK4A expression and cell senescence on human renal cancer cells in vitro, consequently promoting the anticancer activity of calcitriol [59]. Of note, renal cell carcinomas are associated with Von Hippel–Lindau (VHL) disease, an autosomal-dominant disorder caused by germline mutations of the VHL tumor-suppressor gene. Surprisingly, acute VHL inactivation has also been described as a trigger of HIF-independent senescence program mediated by Rb and p400 [60]. Interestingly, decreased p400 expression in renal cell carcinoma patients was associated with advanced tumor stage, higher grade of malignancy, and regional lymph node metastasis [61]. Together, these data suggest that the highly proliferative, decreased-p400 subgroup of renal cell carcinomas represents tumors that are characterized by a loss of senescence, making senescence an efficient tumor-suppressive mechanism that must be overcome to develop VHL-associated neoplasia. Importantly, senescence can also act as a driver of cancer development by altering the cellular microenvironment, mainly through the SMS components [62][63]. However, whether cellular senescence can favor tumor progression in renal cancers remains to be determined.
References
- Lecot, P.; Alimirah, F.; Desprez, P.-Y.; Campisi, J.; Wiley, C. Context-dependent effects of cellular senescence in cancer development. Br. J. Cancer 2016, 114, 1180–1184. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Khosla, S.; Farr, J.N.; Tchkonia, T.; Kirkland, J.L. The role of cellular senescence in ageing and endocrine disease. Nat. Rev. Endocrinol. 2020, 16, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; Fagagna, F. d’Adda di Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef]
- Kuilman, T.; Peeper, D.S. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer 2009, 9, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef]
- Sadaie, M.; Salama, R.; Carroll, T.; Tomimatsu, K.; Chandra, T.; Young, A.R.J.; Narita, M.; Pérez-Mancera, P.A.; Bennett, D.C.; Chong, H.; et al. Redistribution of the Lamin B1 genomic binding profile affects rearrangement of heterochromatic domains and SAHF formation during senescence. Genes Dev. 2013, 27, 1800–1808. [Google Scholar] [CrossRef]
- Itahana, K.; Itahana, Y.; Dimri, G.P. Colorimetric Detection of Senescence-Associated β Galactosidase. Methods Mol. Biol. 2013, 965, 143–156. [Google Scholar] [CrossRef]
- Georgakopoulou, E.A.; Tsimaratou, K.; Evangelou, K.; Fernandez Marcos, P.J.; Zoumpourlis, V.; Trougakos, I.P.; Kletsas, D.; Bartek, J.; Serrano, M.; Gorgoulis, V.G. Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging 2013, 5, 37–50. [Google Scholar] [CrossRef]
- Dörr, J.R.; Yu, Y.; Milanovic, M.; Beuster, G.; Zasada, C.; Däbritz, J.H.M.; Lisec, J.; Lenze, D.; Gerhardt, A.; Schleicher, K.; et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 2013, 501, 421–425. [Google Scholar] [CrossRef]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef]
- Korolchuk, V.I.; Miwa, S.; Carroll, B.; von Zglinicki, T. Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? EBioMedicine 2017, 21, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Bakula, D.; Scheibye-Knudsen, M. MitophAging: Mitophagy in Aging and Disease. Front. Cell Dev. Biol. 2020, 8, 239. [Google Scholar] [CrossRef]
- Tchkonia, T.; Kirkland, J.L. Aging, Cell Senescence, and Chronic Disease: Emerging Therapeutic Strategies. JAMA 2018, 320, 1319–1320. [Google Scholar] [CrossRef]
- Palmer, A.K.; Xu, M.; Zhu, Y.; Pirtskhalava, T.; Weivoda, M.M.; Hachfeld, C.M.; Prata, L.G.; van Dijk, T.H.; Verkade, E.; Casaclang-Verzosa, G.; et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 2019, 18, e12950. [Google Scholar] [CrossRef]
- Barnes, P.J.; Baker, J.; Donnelly, L.E. Cellular Senescence as a Mechanism and Target in Chronic Lung Diseases. Am. J. Respir. Crit. Care Med. 2019, 200, 556–564. [Google Scholar] [CrossRef]
- Song, P.; Zhao, Q.; Zou, M.-H. Targeting senescent cells to attenuate cardiovascular disease progression. Ageing Res. Rev. 2020, 60, 101072. [Google Scholar] [CrossRef] [PubMed]
- Musi, N.; Valentine, J.M.; Sickora, K.R.; Baeuerle, E.; Thompson, C.S.; Shen, Q.; Orr, M.E. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 2018, 17, e12840. [Google Scholar] [CrossRef]
- Riessland, M.; Kolisnyk, B.; Kim, T.W.; Cheng, J.; Ni, J.; Pearson, J.A.; Park, E.J.; Dam, K.; Acehan, D.; Ramos-Espiritu, L.S.; et al. Loss of SATB1 induces p21-dependent cellular senescence in post-mitotic dopaminergic neurons. Cell Stem Cell 2019, 25, 514–530.e8. [Google Scholar] [CrossRef] [PubMed]
- Berkenkamp, B.; Susnik, N.; Baisantry, A.; Kuznetsova, I.; Jacobi, C.; Sörensen-Zender, I.; Broecker, V.; Haller, H.; Melk, A.; Schmitt, R. In vivo and in vitro analysis of age-associated changes and somatic cellular senescence in renal epithelial cells. PLoS ONE 2014, 9, e88071. [Google Scholar] [CrossRef]
- Melk, A.; Schmidt, B.M.W.; Takeuchi, O.; Sawitzki, B.; Rayner, D.C.; Halloran, P.F. Expression of p16INK4a and other cell cycle regulator and senescence associated genes in aging human kidney. Kidney Int. 2004, 65, 510–520. [Google Scholar] [CrossRef]
- Klee, N.S.; McCarthy, C.G.; Lewis, S.; McKenzie, J.L.; Vincent, J.E.; Webb, R.C. Urothelial Senescence in the Pathophysiology of Diabetic Bladder Dysfunction—A Novel Hypothesis. Front. Surg. 2018, 5, 5. [Google Scholar] [CrossRef]
- Choi, J.; Shendrik, I.; Peacocke, M.; Peehl, D.; Buttyan, R.; Ikeguchi, E.F.; Katz, A.E.; Benson, M.C. Expression of senescence-associated beta-galactosidase in enlarged prostates from men with benign prostatic hyperplasia. Urology 2000, 56, 160–166. [Google Scholar] [CrossRef]
- Sturmlechner, I.; Durik, M.; Sieben, C.J.; Baker, D.J.; van Deursen, J.M. Cellular senescence in renal ageing and disease. Nat. Rev. Nephrol. 2017, 13, 77–89. [Google Scholar] [CrossRef]
- Karam, Z.; Tuazon, J. Anatomic and physiologic changes of the aging kidney. Clin. Geriatr. Med. 2013, 29, 555–564. [Google Scholar] [CrossRef]
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O’Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D.R. Global Prevalence of Chronic Kidney Disease–A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef] [PubMed]
- Goligorsky, M.S. Chronic Kidney Disease: A Vicarious Relation to Premature Cell Senescence. Am. J. Pathol. 2020, 190, 1164–1171. [Google Scholar] [CrossRef]
- Zhou, B.; Wan, Y.; Chen, R.; Zhang, C.; Li, X.; Meng, F.; Glaser, S.; Wu, N.; Zhou, T.; Li, S.; et al. The emerging role of cellular senescence in renal diseases. J. Cell. Mol. Med. 2020, 24, 2087–2097. [Google Scholar] [CrossRef]
- Wang, W.-J.; Cai, G.-Y.; Chen, X.-M. Cellular senescence, senescence-associated secretory phenotype, and chronic kidney disease. Oncotarget 2017, 8, 64520–64533. [Google Scholar] [CrossRef]
- Schmitt, R.; Coca, S.; Kanbay, M.; Tinetti, M.E.; Cantley, L.G.; Parikh, C.R. Recovery of kidney function after acute kidney injury in the elderly: A systematic review and meta-analysis. Am. J. Kidney Dis. 2008, 52, 262–271. [Google Scholar] [CrossRef]
- Sörensen-Zender, I.; Rong, S.; Susnik, N.; Zender, S.; Pennekamp, P.; Melk, A.; Haller, H.; Schmitt, R. Renal tubular Notch signaling triggers a prosenescent state after acute kidney injury. Am. J. Physiol. Ren. Physiol. 2014, 306, F907–F915. [Google Scholar] [CrossRef]
- Cheng, H.; Fan, X.; Lawson, W.E.; Paueksakon, P.; Harris, R.C. Telomerase deficiency delays renal recovery in mice after ischemia reperfusion injury by impairing autophagy. Kidney Int. 2015, 88, 85–94. [Google Scholar] [CrossRef]
- Lee, D.H.; Wolstein, J.M.; Pudasaini, B.; Plotkin, M. INK4a deletion results in improved kidney regeneration and decreased capillary rarefaction after ischemia-reperfusion injury. Am. J. Physiol. Ren. Physiol. 2011, 302, F183–F191. [Google Scholar] [CrossRef] [PubMed]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef]
- Megyesi, J.; Andrade, L.; Vieira, J.M.; Safirstein, R.L.; Price, P.M. Positive effect of the induction of p21WAF1/CIP1 on the course of ischemic acute renal failure. Kidney Int. 2001, 60, 2164–2172. [Google Scholar] [CrossRef]
- DiRocco, D.P.; Bisi, J.; Roberts, P.; Strum, J.; Wong, K.-K.; Sharpless, N.; Humphreys, B.D. CDK4/6 inhibition induces epithelial cell cycle arrest and ameliorates acute kidney injury. Am. J. Physiol. Ren. Physiol. 2014, 306, F379–F388. [Google Scholar] [CrossRef] [PubMed]
- Verzola, D.; Gandolfo, M.T.; Gaetani, G.; Ferraris, A.; Mangerini, R.; Ferrario, F.; Villaggio, B.; Gianiorio, F.; Tosetti, F.; Weiss, U.; et al. Accelerated senescence in the kidneys of patients with type 2 diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2008, 295, F1563–F1573. [Google Scholar] [CrossRef] [PubMed]
- Kitada, K.; Nakano, D.; Ohsaki, H.; Hitomi, H.; Minamino, T.; Yatabe, J.; Felder, R.A.; Mori, H.; Masaki, T.; Kobori, H.; et al. Hyperglycemia causes cellular senescence via a SGLT2- and p21-dependent pathway in proximal tubules in the early stage of diabetic nephropathy. J. Diabetes Complicat. 2014, 28, 604–611. [Google Scholar] [CrossRef]
- Prattichizzo, F.; De Nigris, V.; Mancuso, E.; Spiga, R.; Giuliani, A.; Matacchione, G.; Lazzarini, R.; Marcheselli, F.; Recchioni, R.; Testa, R.; et al. Short-term sustained hyperglycaemia fosters an archetypal senescence-associated secretory phenotype in endothelial cells and macrophages. Redox Biol. 2017, 15, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Perlman, A.S.; Chevalier, J.M.; Wilkinson, P.; Liu, H.; Parker, T.; Levine, D.M.; Sloan, B.J.; Gong, A.; Sherman, R.; Farrell, F.X. Serum Inflammatory and Immune Mediators Are Elevated in Early Stage Diabetic Nephropathy. Ann. Clin. Lab. Sci. 2015, 45, 256–263. [Google Scholar] [PubMed]
- Lujambio, A. To clear, or not to clear (senescent cells)? That is the question. Bioessays 2016, 38 (Suppl. 1), S56–S64. [Google Scholar] [CrossRef] [PubMed]
- Clements, M.E.; Chaber, C.J.; Ledbetter, S.R.; Zuk, A. Increased Cellular Senescence and Vascular Rarefaction Exacerbate the Progression of Kidney Fibrosis in Aged Mice Following Transient Ischemic Injury. PLoS ONE 2013, 8, e70464. [Google Scholar] [CrossRef]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Zhou, S.; Zhou, Z.; Liu, Y.; Yang, L.; Liu, J.; Zhang, Y.; Li, H.; Liu, Y.; Hou, F.F.; et al. Wnt9a Promotes Renal Fibrosis by Accelerating Cellular Senescence in Tubular Epithelial Cells. JASN 2018, 29, 1238–1256. [Google Scholar] [CrossRef]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef]
- Wolstein, J.M.; Lee, D.H.; Michaud, J.; Buot, V.; Stefanchik, B.; Plotkin, M.D. INK4a knockout mice exhibit increased fibrosis under normal conditions and in response to unilateral ureteral obstruction. Am. J. Physiol. Ren. Physiol. 2010, 299, F1486–F1495. [Google Scholar] [CrossRef]
- Melk, A.; Schmidt, B.M.W.; Vongwiwatana, A.; Rayner, D.C.; Halloran, P.F. Increased expression of senescence-associated cell cycle inhibitor p16INK4a in deteriorating renal transplants and diseased native kidney. Am. J. Transplant. 2005, 5, 1375–1382. [Google Scholar] [CrossRef]
- Ferlicot, S.; Durrbach, A.; Bâ, N.; Desvaux, D.; Bedossa, P.; Paradis, V. The role of replicative senescence in chronic allograft nephropathy. Hum. Pathol. 2003, 34, 924–928. [Google Scholar] [CrossRef]
- Joosten, S.A.; van Ham, V.; Nolan, C.E.; Borrias, M.C.; Jardine, A.G.; Shiels, P.G.; van Kooten, C.; Paul, L.C. Telomere Shortening and Cellular Senescence in a Model of Chronic Renal Allograft Rejection. Am. J. Pathol. 2003, 162, 1305–1312. [Google Scholar] [CrossRef]
- Braun, H.; Schmidt, B.M.W.; Raiss, M.; Baisantry, A.; Mircea-Constantin, D.; Wang, S.; Gross, M.-L.; Serrano, M.; Schmitt, R.; Melk, A. Cellular Senescence Limits Regenerative Capacity and Allograft Survival. JASN 2012, 23, 1467–1473. [Google Scholar] [CrossRef]
- Domański, L.; Kłoda, K.; Kwiatkowska, E.; Borowiecka, E.; Safranow, K.; Drozd, A.; Ciechanowicz, A.; Ciechanowski, K. Effect of delayed graft function, acute rejection and chronic allograft dysfunction on kidney allograft telomere length in patients after transplantation: A prospective cohort study. BMC Nephrol. 2015, 16, 23. [Google Scholar] [CrossRef] [PubMed]
- van Willigenburg, H.; de Keizer, P.L.J.; de Bruin, R.W.F. Cellular senescence as a therapeutic target to improve renal transplantation outcome. Pharmacol. Res. 2018, 130, 322–330. [Google Scholar] [CrossRef]
- Melk, A.; Halloran, P.F. Cell Senescence and Its Implications for Nephrology. JASN 2001, 12, 385–393. [Google Scholar]
- McGlynn, L.M.; Stevenson, K.; Lamb, K.; Zino, S.; Brown, M.; Prina, A.; Kingsmore, D.; Shiels, P.G. Cellular senescence in pretransplant renal biopsies predicts postoperative organ function. Aging Cell 2009, 8, 45–51. [Google Scholar] [CrossRef]
- Lee, S.; Lee, J.-S. Cellular senescence: A promising strategy for cancer therapy. BMB Rep. 2019, 52, 35–41. [Google Scholar] [CrossRef]
- Cole, A.M.; Ridgway, R.A.; Derkits, S.E.; Parry, L.; Barker, N.; Clevers, H.; Clarke, A.R.; Sansom, O.J. p21 loss blocks senescence following Apc loss and provokes tumourigenesis in the renal but not the intestinal epithelium. EMBO Mol. Med. 2010, 2, 472–486. [Google Scholar] [CrossRef]
- Shen, Y.; Yu, D.; Qi, P.; Wang, X.; Guo, X.; Zhang, A. Calcitriol induces cell senescence of kidney cancer through JMJD3 mediated histone demethylation. Oncotarget 2017, 8, 100187–100195. [Google Scholar] [CrossRef] [PubMed]
- Young, A.P.; Schlisio, S.; Minamishima, Y.A.; Zhang, Q.; Li, L.; Grisanzio, C.; Signoretti, S.; Kaelin, W.G. VHL loss actuates a HIF-independent senescence programme mediated by Rb and p400. Nat. Cell Biol. 2008, 10, 361–369. [Google Scholar] [CrossRef]
- Macher-Goeppinger, S.; Bermejo, J.L.; Schirmacher, P.; Pahernik, S.; Hohenfellner, M.; Roth, W. Senescence-associated protein p400 is a prognostic marker in renal cell carcinoma. Oncol. Rep. 2013, 30, 2245–2253. [Google Scholar] [CrossRef]
- Schosserer, M.; Grillari, J.; Breitenbach, M. The Dual Role of Cellular Senescence in Developing Tumors and Their Response to Cancer Therapy. Front. Oncol. 2017, 7, 278. [Google Scholar] [CrossRef]
- Zeng, S.; Shen, W.H.; Liu, L. Senescence and Cancer. Cancer Transl. Med. 2018, 4, 70–74.



