2. General Anti-Tumor Molecular Mechanisms of OVs
2.1. OVs Lyse Tumors Directly but Not Ordinary Cells
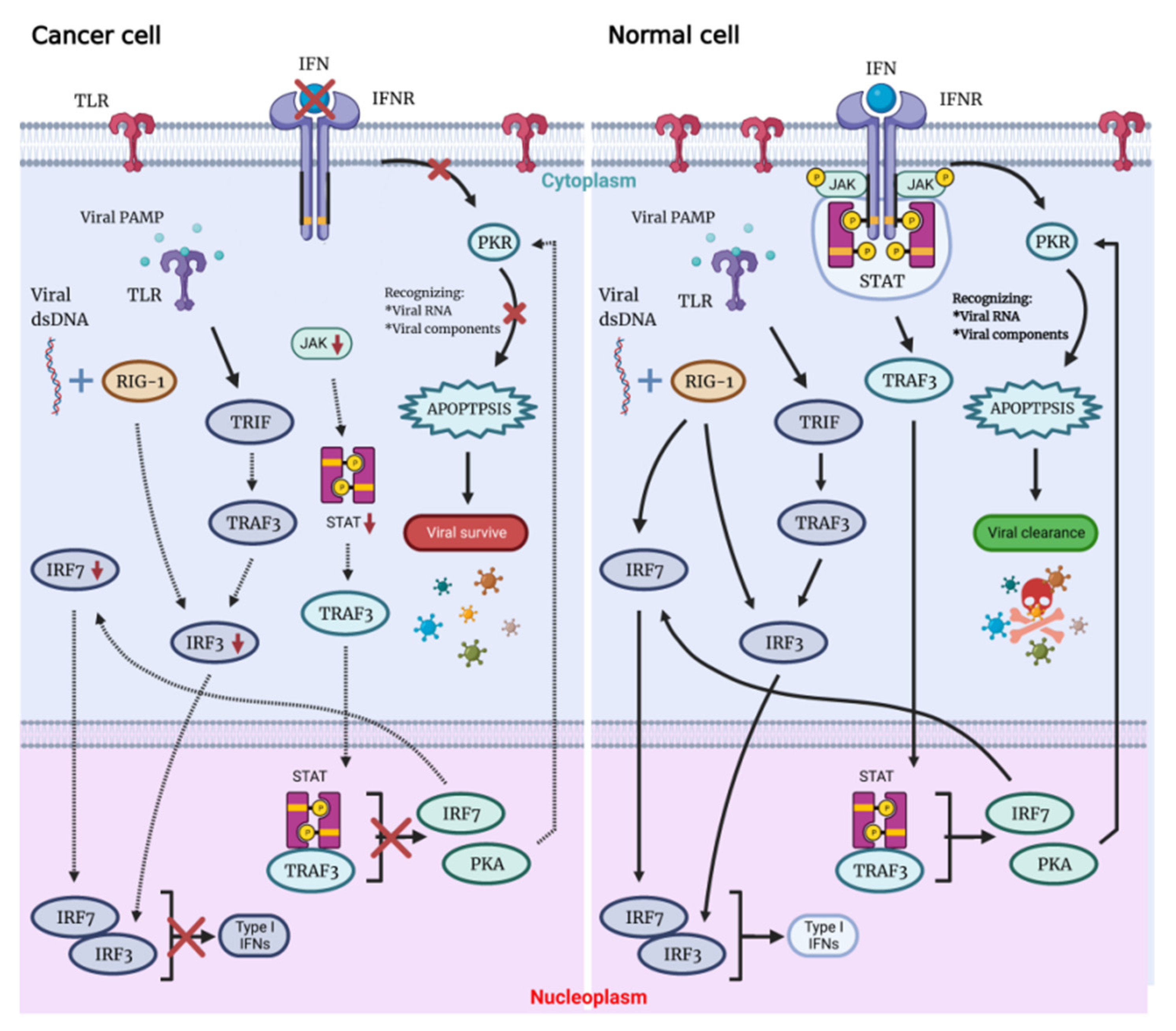
Possessing the ability to replicate through the lytic cycle, OVs can directly destroy the tumor cells of the host and infect neighboring cells with newly formed virions. Not only associated with cellular antiviral response components (PKR, Toll-like receptor TLR, retinoic acid-inducible gene 1 RIG-1, IFN, etc.), this capacity also depends on virus type, dose, natural and induced virulence, and susceptibility of cancer cells to different forms of cell death (apoptosis, necrosis, cytokinesis and autophagy). On the contrary, there are multiple signaling pathways that act to detect and clear viral particles for normal cells. Defects in these pathways in tumor cells are key to their inability to clear the virus and be lysed by the virus (
Figure 1)
[6].
Figure 1. The OVs can specifically lyse tumor cells, sparing normal cells. Following viral infection, most normal cells activate the antiviral pathway, thereby controlling the infection. The antiviral mechanism can be triggered by activating the Toll-like receptor and RIG-I-like receptor signaling by viral PAMPs, which might include viral nucleic acids. Once the virus is detected, a signaling cascade of several IFN components JAK, STAT and interferon regulatory factor 9 (IRF9) leads to a programmed transcription pathway that limits the spread of the virus and can target infected cells for apoptosis or necrosis. Local IFN production induced by the innate immune response to viral infection may also contribute to antiviral activity via IFNR. Type I IFN signals through the JAK-STAT signaling pathway lead to the upregulation of cell cycle regulators, such as PKR and IRF7. These regulators limit viral spread by binding to viral particles and triggering the type I IFN transcriptional pathway, promoting aborted apoptosis and cytokine production in infected cells.
When OV infects normal cells, the viral components trigger antiviral immune response through multiple mechanisms that involve stimulation of the intracellular Toll-like receptors (TLRs), which induce secretion of interferon type I (IFN-I), which triggers antiviral immune response. Expressed in a variety of cells, TLRs are pattern recognition receptors on the cell surface and in cells that could be activated by pathogen-associated molecular patterns (PAMPs), with repetitive sequences common to pathogenic bacteria and viruses. These PAMPs include viral capsids, DNA, RNA and viral proteins. TLR will then activate TNF-related factor 3 (TRAF3). RIG-1 recognizes viral nucleic acids and is activated, which together with TRAF3 continues to activate downstream factors, such as IFN-related factor 3 (IRF3) and IRF7, thereby further activating the JAK-STAT (Janus kinase signal transducer and activator of transcription) pathway, which coordinates the antiviral machinery in infected cells. Factors downstream of this pathway, such as IRF7, enhance local IFN release and promote IFN-mediated antiviral responses. The TLR pathway and IFN pathway are interconnected to trigger the antiviral immune response in normal cells (
Figure 1)
[7][8][9]. In addition, TLR signaling activates dendritic cells (DCs), as well as macrophages Mø to secrete IL-12—a cytokine that induces the conversion of helper T cells to the Th1 phenotype
[10], which plays a role in the activation of anti-tumor immunity by OVs.
By binding to the IFN receptor (IFNR), IFN activates PKR, an intracellular protein kinase that recognizes double-stranded RNA and other viral components. When PKR is activated, it terminates cellular protein synthesis and promotes rapid cell death and virus clearance
[11][12]. Moreover, IFN signaling through the IFNAR receptor ultimately leads to the expression of interferon-stimulated genes (ISG). When viral proteins are modified by ISG, their own localization and protease activity are affected, oligomerization and geometry are disrupted, and interactions with host proteins or other viral proteins are destroyed, which eventually leads to the reduction of virus replication.
Immune deficiency is often present in tumor cells, meaning that abnormalities in the IFN pathway and PKR activity can interfere with viral clearance, with rapid viral replication and lysis of host cells, resulting in specific killing of tumor cells by OVs. OVs can manipulate various signaling pathways within tumor cells to prevent apoptosis, giving the viruses more time to complete their life cycle. OVs ultimately induce cell death, and both the form of cell death and the release of danger signals from virally infected cells can greatly assist in the induction of a host immune response, which not only directly clears tumor cells but also sets the stage for the initiation of a systemic immune response.
2.2. OVs Induce Anti-Tumor Immunity
2.2.1. OVs Induce Local and Systemic Anti-Tumor Immunity
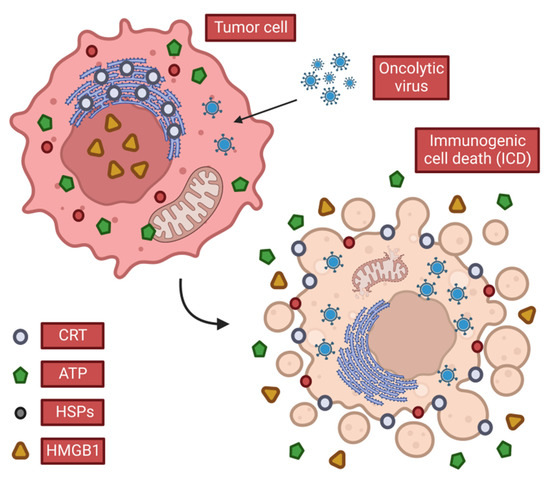
Possessing a broad ability to activate the body′s immune system, OVs can cause immunogenic cell death (ICD) of tumor cells (
Figure 2) by lysing cells, releasing soluble antigens, danger signals and IFN thereby stimulating anti-tumor immunity
[13]. OVs also induce T cell responses in tumors, which can promote local inflammation and recruit CD4
+ and CD8
+ T cells associated with an anti-tumor response
[14][15][16]. In the meantime, the “neoantigens” that are transiently expressed during normal tissue repair after local inflammation induce immune surveillance-related immune responses, which also help clear tumor cells that express “neoantigens” but are not infected with the virus. After tumor cells are lysed, the local release of cytotoxic perforin and granzyme may kill nearby tumor cells, whether they are infected by viruses or not. This phenomenon of local adjacent tumor cell death through immune inflammation and other means is called the bystander killing effect. This induction of local, systemic innate, and tumor-specific immune responses appears to be a key factor in tumor eradication by OVs, as opposed to the direct lysis of tumor cells.
Figure 2. Characteristics of immunogenic cell death. When OVs cause ICDs in tumor cells, they release or expose DAMPs that stimulate anti-tumor immune responses. Tumor cells release ATP and HMGB1 in the extracellular space and expose CRT and heat shock protein (HSP), which promotes the continuation of anti-tumor immunity.
When cells infected with OVs die, they release tumor-associated antigens (TAAs), viral PAMPS, and cellular damage-associated molecular patterns DAMPs (e.g., heat shock proteins, high mobility group B1 (HMGB1), calreticulin (CALR), ATP and uric acid, etc.) and cytokines (such as type I IFN, TNF-α, IFN-γ and IL-12). Among these, CALR, ATP and HMGB1 are considered to play a key role among all three DAMPs.
CALR
CALR is thought to be transferred to the cell surface early in the ICD process (
Figure 2)
[17][18][19][20][21], and upon binding to low-density lipoprotein receptor-related protein 1 (LRP1, also known as CD91), it transmits phagocytic signals to APCs, such as dendritic cells (DCs), consequently increasing the ability to phagocytize dead cells
[22][23][24][25][26][27][28][29][30][31]. The endoplasmic reticulum stress response is the basis of CALR exposure
[32]. Interestingly, phagocytosis stimulation by CALR is neutralized by CD47 expression in a large number of solid and hematopoietic tumors
[24]. This indicates that tumor cells can avoid the effect of CALR through CD47, so as to prevent immune killing caused by physiologically dead tumor cells via CALR in the process of tumorigenesis.
ATP
ATP released extracellularly by tumor cells following the direct lysis of OVs is a powerful chemoattractant (
Figure 2). It promotes not only the recruitment of immune cells to ICD sites but also the differentiation of these immune cells. These functions are associated with the G-protein coupled purinergic receptor 2 (receptor P2Y, G-protein coupled, 2; P2RY2)
[33][34][35][36]. In addition, extracellular ATP promotes the activation of the NLR family and pyrin-containing structural domain 3 (NLRP3) inflammasome in APCs, thereby stimulating the release of IL-1β and IL-18, which are two significant pro-inflammatory cytokines involved in the immune response
[37][38][39][40][41][42][43][44][45][46][47]. This immune process can interfere if pericellular ATP is converted to ADP or AMP using recombinant adenosine triphosphate bisphosphatase (apyrase, an ATP degrading enzyme) or exonucleoside triphosphate diphosphate hydrolase 1 (ENTPD1, i.e., CD39)
[48]. Similar to the above anti-CALR effect, tumor cells overexpress CD39 and the 5′-nucleotidase ecto (NT5E, i.e., CD73) to convert ATP to adenosine and thus exert immunosuppressive effects to ensure their own growth
[49][50][51][52][53][54].
HMGB1
When HMGB1 is released extracellularly (
Figure 2), it binds to several receptors (TLR2, TLR4, AGER) on the surface of immune cells, mediates strong pro-inflammatory effects
[55][56][57][58][59][60][61][62][63], and can also exert immune chemotactic activity by forming complexes with chemokine (CXC motif) ligand 12 (CXCL12)
[64]. Also, endogenous HMGB1 promotes autophagy by interfering with the reciprocal inhibition between the central sub-temporal regulator beclin 1 (BECN1) and the anti-apoptotic protein B-cell CLL/lymphoma 2 (BCL2)
[65][66][67], so researchers speculate that HMGB1 release may contribute to the ICD inducer-induced cellular autophagic response.
IL-12
In addition to the above three substances (CALR, HMGB1, ATP), interleukin-12 (IL-12), a potent anti-cancer cytokine, is also important in the induction of anti-tumor immunity by OVs. IL-12 is normally produced by phagocytes (monocytes/macrophages, neutrophils) and dendritic cells in response to pathogens and directly activates innate immune cells (NK cells, NK-T cells) and adaptive immune cells (CD4
+ cells, CD8
+ cells). IL-12 initiates T cells and enhances their survival, promotes Th1 differentiation and enhances T cell, NK cell and NK-T cell effector functions. As mentioned earlier, tumor immunotherapy requires the involvement of T cells, while OVs and IL-12 can act synergistically to increase the recruitment of CTL cells into the tumor microenvironment (TME). IL-12 also induces the secretion of IFN-γ, which acts directly on tumor cells in the tumor microenvironment as well as on stromal and endothelial cells, and its signaling leads to (1) increased MHC I processing and presentation (enhancing tumor recognition by T cells) and (2) induction of chemokines IP-10 and MIG, recruiting innate and adaptive immune effectors, leading to (i) altered extracellular matrix remodeling, including inhibition of matrix metalloproteinase expression, which reduces angiogenesis and tumor invasion, and (ii) reduced expression of adhesion molecules in endothelial cells, which may further limit angiogenesis
[68][69][70][71].
These substances promote the maturation of antigen-presenting cells’ APCs (e.g., DCs), which promote the differentiation of T cells towards specific CD4
+ T cells and CD8
+ T cells
[72]. CD8
+ T cells act as cytotoxic T lymphocytes (CTL) that, when activated, metastasize to the site of tumor growth and recognize tumor-specific antigens to mediate anti-tumor immunity
[9]. This process is vital for systemic anti-tumor immunity, and the importance of tumor-specific CD8
+ T cells in mediating immune tumor elimination of OVs has been demonstrated in many pre-clinical studies
[16][73][74][75][76]. The type I IFN and DAMP released by OVs after tumor lysis also mediate the activation of natural killer (NK) cells, further promoting an anti-tumor immune response as part of innate immunity
[77]. Moreover, OV infection leads to downregulation of major histocompatibility complex (MHC) class I expression on the host cell surface
[78][79], and NK cells can kill target cells with downregulated MHC class I expression, which is common in cancer cells
[80][81]. Notably, IFN-γ released from tumor cells by OVs upregulates the expression of MHC class I on the surface of nearby cancer cells, thereby enhancing the CTL-mediated immune response (
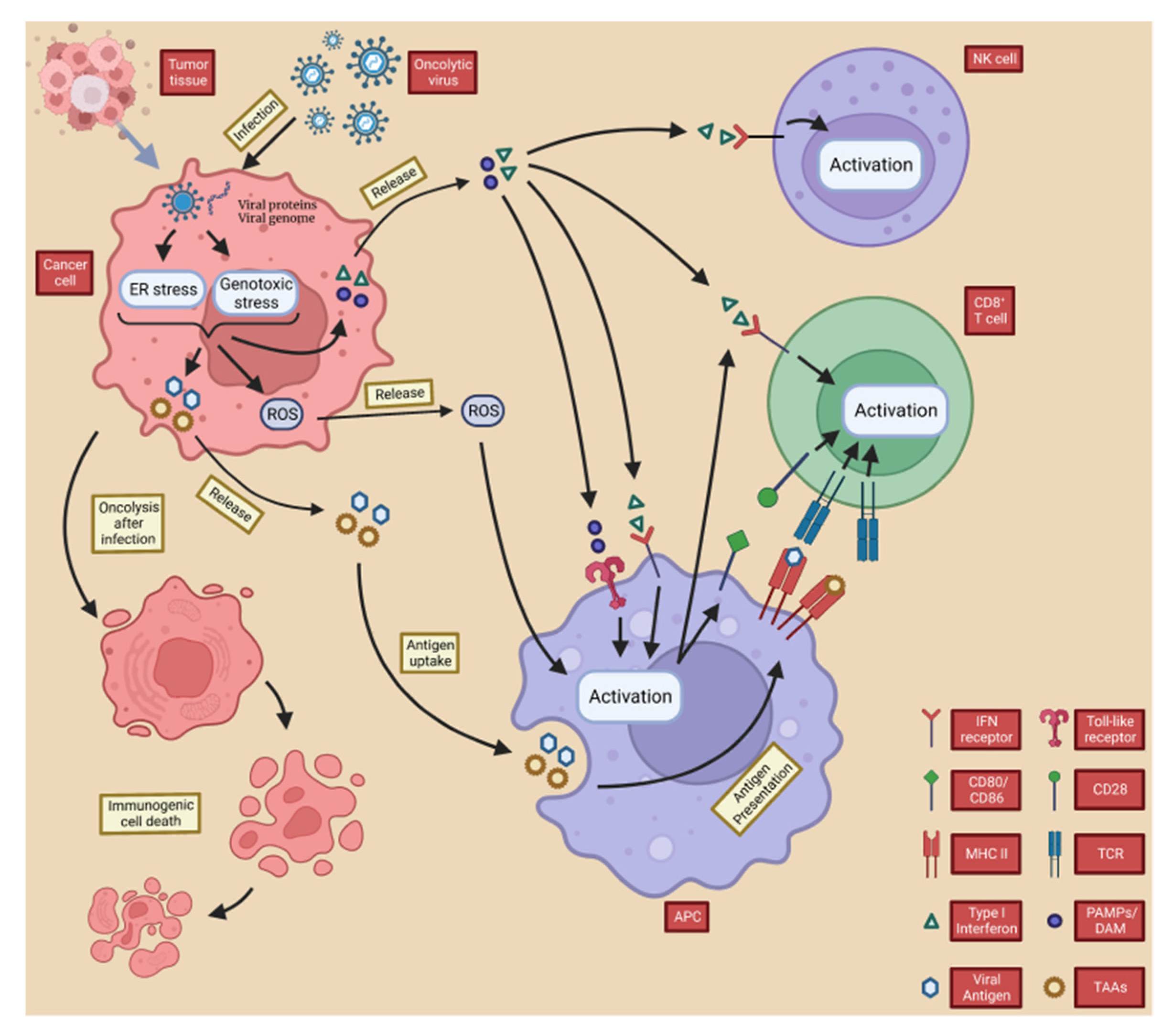
Figure 3)
[82][83][84][85].
Figure 3. OVs induce local and systemic anti-tumor immunity. Following infection with the oncolytic virus, cancer cells initiate an antiviral response consisting of endoplasmic reticulum (ER) and genotoxic stresses, which results in the upregulation of reactive oxygen species (ROS) and the production of antiviral cytokines. ROS and cytokines, particularly type I interferon (IFN), are released from infected cancer cells and stimulate immune cells (antigen-presenting cells, CD8+ T cells, and NK cells). PAMP (made up of viral particles) and DAMP (made up of host cell proteins) stimulate the immune system by triggering activation receptors, such as TLRs. In the resulting immune-stimulatory environment, TAA and neoantigens are taken up and released by antigen-presenting cells.
Overall, TAA, PAMPS, DAMP and cytokines released by OVs from directly lysed cells not only cause ICD in local and distant tumor cells by inducing inflammation and activating adaptive T cells, but also promote the recruitment of APCs to the site of ongoing ICD, triggering adaptive immunity to further enhance ICD and creating positive feedback
[86]. These actions lead to changes in the composition of the TME, accordingly disrupting the original tumor cell growth environment and laying the foundation for long-term cancer eradication.
2.2.2. OVs Counteract Tumor Immune Evasion
Tumor immune evasion is considered one of the “hallmarks of cancer” and represents a major direction for new cancer therapy
[87]. Cancer cells avoid the destruction of immune-mediated responses through complex mechanisms, including the progressive development of an immunosuppressive environment within the tumor and the selection of tumor variants against immune effectors (sometimes called “immunoediting”)
[88]. There are currently six known mechanisms of immune evasion: (1) Fas/Fasl-mediated immune evasion; (2) decreased immunogenicity and antigen modulation of tumor-associated antigens; (3) decreased or absent expression of MHC molecules on tumor cell surfaces; (4) expression of immunosuppressive molecules, such as PD-L1 and CTLA-4 on the surface of tumor cells to disable T cells; (5) release of tumor antigen molecules that bind to antibodies, or the NK cells and macrophages through the FC segment of antibodies to block the ADCC effect; and(6) tumor-mediated autocrine or paracrine production of immunosuppressive cytokines, such as IL-10, transforming growth factor-β (TGF-β) and indoleamine-2,3-Dioxygenase (IDO) to promote the recruitment of immunosuppressive cells, such as tumor-associated macrophages and myeloid-derived suppressor cells (MDSCs), to tumor growth sites
[89][90][91][92][93]. Limited circulating immune effector cell infiltration is also characteristic of the immunosuppressive tumor microenvironment. Overall, the mechanism by which OVs counteract tumor immune evasion is via poor T-cell infiltration, low tumor mutational burden (TMB), and the establishment of a non-immunogenic tumor microenvironment
[94].
Remarkably, based on the spatial distribution of CTLs in the tumor microenvironment (TME), tumors can be classified into three basic immunophenotypes: immune inflammation, immune rejection and immune desert. The immune inflammation phenotype, also known as the immunogenic phenotype, is termed a “hot” tumor and responds well to immunotherapy. The immune rejection and immune desert phenotypes are collectively referred to as non-immunogenic phenotypes, which are also known as “cold” tumors and are generally less responsive to immunotherapy
[95][96][97]. Although therapeutic monoclonal antibodies (mAb) against immune checkpoints (ICP), such as anti-PD-1/PD-L1 agents have offered a new approach to tumor therapy recently, immune checkpoint inhibitors (ICI) generally have a limited impact on tumors because of the low level of T cell infiltration within the tumor immune microenvironment (TIME).
OVs restrict the immunosuppressive tumor microenvironment by modifying various mechanisms that alter the cytokine environment and immune cell types
[98][99]. As previously described, it has been shown that upregulation of peripheral cellular MHC and costimulatory molecules can be observed in tumor cells following oncolytic virus infection. This upregulation promotes immune-mediated tumor cell recognition and eradication and triggers tumor-associated antigen exposure and epitope spreading, which in turn recruits immune cell populations and transforms ‘cold’ tumors with a ‘non-T-cell inflammatory’ phenotype into ‘hot’ tumors with a ‘T-cell inflammatory phenotype’
[100][101]. Furthermore, the expression of multiple immunostimulatory transgenes within the recombinant OV genome may reshape the tumor immune microenvironment with minimal toxicity through direct access to the tumor niche. One such transgene is the granulocyte-macrophage colony-stimulating factor (GM-CSF), an immunostimulatory molecule that recruits specialized APCs, including DCs, promotes cancer antigen presentation, recruits immune cells to mature, and activates NK cells and tumor antigen-specific T cells. Simultaneously, the combination of the release of “neoantigens” after the tumor cells are killed and the bystander-killing effect will also “heat up” the tumor immune microenvironment and thus better counter tumor immune evasion
[94][95][96][97].
OV-induced alteration of the tumor immune microenvironment was observed not only at the site of virus injection but also in non-virally injected lesions, suggesting that the mechanism of countering tumor immune evasion can be systemic and can be the subsequent part of OVs inducing local and anti-tumor immune responses to kill tumor cells. Studies have shown that this alteration enhances the effect of ICIs, such as anti-CTLA-4mAb, anti-PD1/PD-L1mAb
[16][102][103][104][105], and that both OVs and ICIs act on the majority of tumors without targeting specific cancer types, suggesting the potential of OVs to be used in combination with tumor immunotherapy.
3. Epidemiology and Current Therapy of PCa
3.1. Global Epidemiology of PCa
Effective PCa treatment is a hot topic of interest to urologists around the world. Although the new coronavirus (COVID-19) has ranked first in deaths from all types of diseases since its globalization in 2019, cancer is still one of the major public health problems worldwide. Globally, PCa is the second most common solid tumor in men, with approximately 13 million new cases diagnosed worldwide each year. According to American Cancer Society statistics in 2022, PCA ranks first in frequency of occurrence (27% of all new cancer cases) and second in number of deaths (11% of 325 all cancer deaths) among men in the United States, which means that PCa remains the most prevalent tumor in the genitourinary system
[106][107]. The incidence of PCa varies considerably between continents and Asia is traditionally considered to be a low incidence region
[108]. Nonetheless, due to economic development, increased life expectancy and westernized lifestyles, the incidence of PCa in Asia is rising rapidly
[109][110]. With almost no symptoms at first, PCa is difficult to detect in its early stages, and at the time of diagnosis, the cancer may already be metastasized and in a terminal stage. According to statistics, around 10 million men are currently diagnosed with PCa, 700,000 of whom have metastases, and the death rate is expected to more than double by 2040
[111][112][113]. PCa that is in the terminal stage have usually already metastasized to, e.g., bone or present as castration-resistant cancer form (CRPC), often difficult to treat.
The median overall survival (OS) for patients with mCRPC ranges from 13 to 32 months, with a 5-year survival rate of 15%. Therefore, the development of new treatments for PCa, especially for intermediate and advanced PCa, is essential.
3.2. Overview of the Current State of PCa Therapy
Treatment options for PCa include active surveillance (AS) and watchful waiting, surgery, endocrine therapy, chemotherapy, radiotherapy, immunotherapy, etc. Summarizing the 2020 edition of European Urology and the guidelines of the National Comprehensive Cancer Network (NCCN)
[114][115][116], as for the traditional treatment, it is now considered that ① active surveillance (AS) as an assessment tool using PSA, DRE, biopsy and other indicators can be applied to patients with low-risk PCa (tumors confined to the peritoneum) to reduce overtreatment and not compromise opportunities for cure. ②Radiotherapy is indicated for low-risk PCa patients with a prostate volume of <50 mL, or in combination with ADT for patients with intermediate or high-risk PCa. ③ Patients with PCa whose tumors are removable and not invading the urethral sphincter have the opportunity to undergo RP, but for patients with intermediate risk, high risk and locally advanced PCa, postoperative adjuvant therapy is also required. ④ Endocrine therapy (androgen deprivation therapy, ADT) is indicated as adjuvant therapy after RP and in combination with the chemotherapeutic agent doxorubicin for metastatic prostate cancer (mPCa), castration-resistant PCa (CRPC) and mCRPC. It has been shown that sex hormones play an important role in the pathogenesis and progression of PCa and that tumor progression can be better controlled after castration
[117].
As for the new treatment, ⑤ the radiopharmaceutical radium 223 is mainly used as a palliative treatment for bone pain in patients with bone metastases and although it is effective in relieving pain, it has no impact on survival
[118]. ⑥ As for targeted drugs, the PARP inhibitors olaparib and rucaparib have also been approved for the treatment of mCRPC in patients with mutations in DNA homologous recombination repair genes (e.g., BRCA1, BRCA2, ATM, PALB2, FANCA, RAD51D, CHEK2)
[119][120][121][122][123][124][125][126][127][128][129][130][131][132][133]. ⑦ For immunotherapy, Sipuleucel-T (a dendritic cell-based autologous vaccine) is thought to increase OS in asymptomatic or mildly symptomatic mCRPC patients. However, due to doubts about its efficacy, manufacturing difficulties and associated costs, this treatment has only been approved by the FDA and not by any European regulatory body, so its use in the clinical setting remains relatively limited
[118]. ⑧ With the rise of ICI therapies in recent years, anti-PD-1 antibody (pembrolizumab), which has been classified as a Class 2B recommendation, has been used to treat advanced PCa with high microsatellite instability (MSI-H)/defective mismatch repair (dMMR)
[134][135][136][137][138][139][140][141][142][143][144].
Current treatment for patients with low-risk PCa is satisfying, but some subpopulations of tumor cells in progressive PCa can alter their nuclear androgen receptor expression levels and function through androgen receptor (AR) increase and hypersensitivity, AR mutations, co-activator/co-inhibitor mutations, androgen non-dependent AR activation and intratumoral androgen production, allowing cancer to survive with low androgen levels or in the absence of androgens (i.e., as CRPC)
[145], making conventional therapies, particularly ADT, less effective. At this time, anti-tumor immunotherapy may be a superior alternative. Immunotherapy for tumors aims to enhance natural defenses to eliminate malignant cells or impair their phenotype and function in the long term, representing a major breakthrough in cancer treatment.
Anti-tumor immunotherapy has long been in the PCa treatment guidelines
[114][115][116], but unfortunately, immune checkpoint inhibitors have not yet shown efficacy in PCa, despite altering clinical outcomes in other solid tumors. Ipilimumab, an anti-cytotoxic T-lymphocyte-associated 4 (CTLA4) checkpoint inhibitor, was studied in two phase III clinical trials of mCRPC, both of which showed no improvement in OS in mCRPC patients
[146][147]. Pembrolizumab showed high response rates in tumors with mismatch repair defects, leading to FDA approval but did not have PCa specificity. Actually, some studies have shown that only 2–12% of PCa have microsatellite instability and hypermutation status that qualify for anti-PD-1 antibody therapy
[148][149]. At the same time, the EAU notes that all patients who receive treatment for mCRPC will eventually progress, which suggests that current therapies are not able to provide a satisfactory survival benefit for mCRPC patients—the median overall survival (OS) for mCRPC patients ranges from 13–32 months, with a 5-year survival rate of 15%. In general, PCa in metastatic form, e.g., with bone metastases in intermediate or advanced stages, or in castration-resistant form (CRPC), is difficult to cure. Therefore, the development of new treatments for advanced PCa, especially more effective anti-tumor immunotherapy, is essential.
 +1 credit
+1 credit