Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Daiki Hayashi | -- | 1784 | 2022-10-25 10:17:27 | | | |
| 2 | Vivi Li | Meta information modification | 1784 | 2022-10-27 04:02:20 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Hayashi, D.; Shirai, Y. Diacylglycerol Kinase in the Amelioration of Diabetic Nephropathy. Encyclopedia. Available online: https://encyclopedia.pub/entry/31310 (accessed on 27 July 2026).
Hayashi D, Shirai Y. Diacylglycerol Kinase in the Amelioration of Diabetic Nephropathy. Encyclopedia. Available at: https://encyclopedia.pub/entry/31310. Accessed July 27, 2026.
Hayashi, Daiki, Yasuhito Shirai. "Diacylglycerol Kinase in the Amelioration of Diabetic Nephropathy" Encyclopedia, https://encyclopedia.pub/entry/31310 (accessed July 27, 2026).
Hayashi, D., & Shirai, Y. (2022, October 26). Diacylglycerol Kinase in the Amelioration of Diabetic Nephropathy. In Encyclopedia. https://encyclopedia.pub/entry/31310
Hayashi, Daiki and Yasuhito Shirai. "Diacylglycerol Kinase in the Amelioration of Diabetic Nephropathy." Encyclopedia. Web. 26 October, 2022.
Copy Citation
The drastic increase in the number of patients with diabetes and its complications is a global issue. Diabetic nephropathy, the leading cause of chronic kidney disease, significantly affects patients’ quality of life and medical expenses. Furthermore, there are limited drugs for treating diabetic nephropathy patients. Impaired lipid signaling, especially abnormal protein kinase C (PKC) activation by de novo-synthesized diacylglycerol (DG) under high blood glucose, is one of the causes of diabetic nephropathy. DG kinase (DGK) is an enzyme that phosphorylates DG and generates phosphatidic acid, i.e., DGK can inhibit PKC activation under diabetic conditions. Indeed, it has been proven that DGK activation ameliorates diabetic nephropathy.
diabetic nephropathy
protein kinase C
diacylglycerol kinase
67 kDa laminin receptor
1. Diabetes and Diabetic Nephropathy
Diabetes is a chronic metabolic disease that causes high blood glucose levels, resulting in serious health problems and death. In 2021, the number of people living with diabetes globally was estimated to be 536.6 million (prevalence: 10.5%), and this number is expected to reach 783.2 million (prevalence: 12.2%) by 2045 [1]. Diabetes significantly increases the risk of various health issues such as heart attacks, strokes, and infections compared to healthy individuals [2][3]. Diabetes also causes serious vascular complications. Diabetic retinopathy, neuropathy, and nephropathy are major microvascular complications of diabetes caused by microangiopathy. These complications lead to blindness, gangrene, and end-stage renal disease, respectively. Diabetic nephropathy (DN) is the leading cause of chronic kidney disease (CKD) in the world, leading to the need for dialysis, reducing patients’ quality of life, and increasing medical care expenses.
DN causes glomerular filtration failure that leads to uremia in one in three diabetic patients after several years of latency [4]. Since a high blood glucose level is the fundamental cause of the complications, delaying the onset of DN by controlling the blood glucose level is the primary treatment strategy [5]. Recently, many drugs to control blood glucose levels, such as insulin, sodium-glucose co-transporter-2 (SGLT-2) inhibitors, and glucagon-like peptide-1 (GLP-1) receptor agonists, have become available. It is also well recognized that the renin-angiotensin-aldosterone system (RAAS), which controls arterial pressure, plays a pivotal role in the pathogenesis of DN. So far, drugs targeting the RAAS, such as angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs), have been the first choice for preventing and delaying the onset of DN. However, there are limits to the drugs available for DN, and there are no specific therapeutics for ameliorating DN once hyperglycemia and high blood pressure become uncontrolled. Although controlling blood glucose levels is the most simple and effective approach to delaying the onset of microvascular complications, and inhibiting RAAS is effective for DN, finding a novel target with a renoprotective effect was necessary. Therefore, to develop a novel drug for DN, it was vital to understand the mechanisms of the development and progression of DN and find an effective therapeutic target.
2. Hyperglycemia Impairs Lipid Signaling
Among the causes of DN, impaired lipid signaling, especially diacylglycerol (DG) signaling, is well known. Generally, DG is produced from phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] and phosphatidylcholine (PC) by the action of phosphoinositide-specific phospholipase C (PI-PLC) and PC-specific PLC (PC-PLC) upon various growth factors and T-cell receptors (TCR) (Figure 1) [6][7][8]. In addition, DG is generated from the dephosphorylation of phosphatidic acid (PA) by phosphatidate phosphohydrolases (PAPs), including lipins [9][10]. PA is generated by hydrolyzing PC by phospholipase D (PLD) [11] and de novo synthesis from glycerol [12]. Glyceraldehyde 3-phosphate, an intermediate of glycolysis, is reversibly isomerized into dihydroxyacetone-phosphate and produces glycerol 3-phosphate. The glycerol 3-phosphate undergoes acylation and generates phosphatidic acid (PA), and dephosphorylates PA into DG (Figure 1). Under hyperglycemic conditions, excess blood glucose is uptaken intracellularly by the glucose transporter (GLUT) [13][14], and excess intracellular glucose results in DG production by de novo synthesis [12]. Both DG and PA are important intermediates for membrane lipids and are bioactive lipids involved in various signal transductions. For example, DG regulates conventional and novel protein kinase C (cPKC and nPKC) [15][16], Ras guanyl nucleotide-releasing protein (RasGRP) [17], and transient receptor potential channel (TRPC) [18]. PA regulates atypical PKC (aPKC) [19], phosphatidylinositol-4-phosphate 5-kinase (PI4P5K) [20], Raf-1 kinase [21], and mammalian target of rapamycin (mTOR) [22]. Therefore, abnormal DG production causes severe changes in their signaling pathways. The abnormal activation of cPKC and nPKC has been reported as a cause of DN.
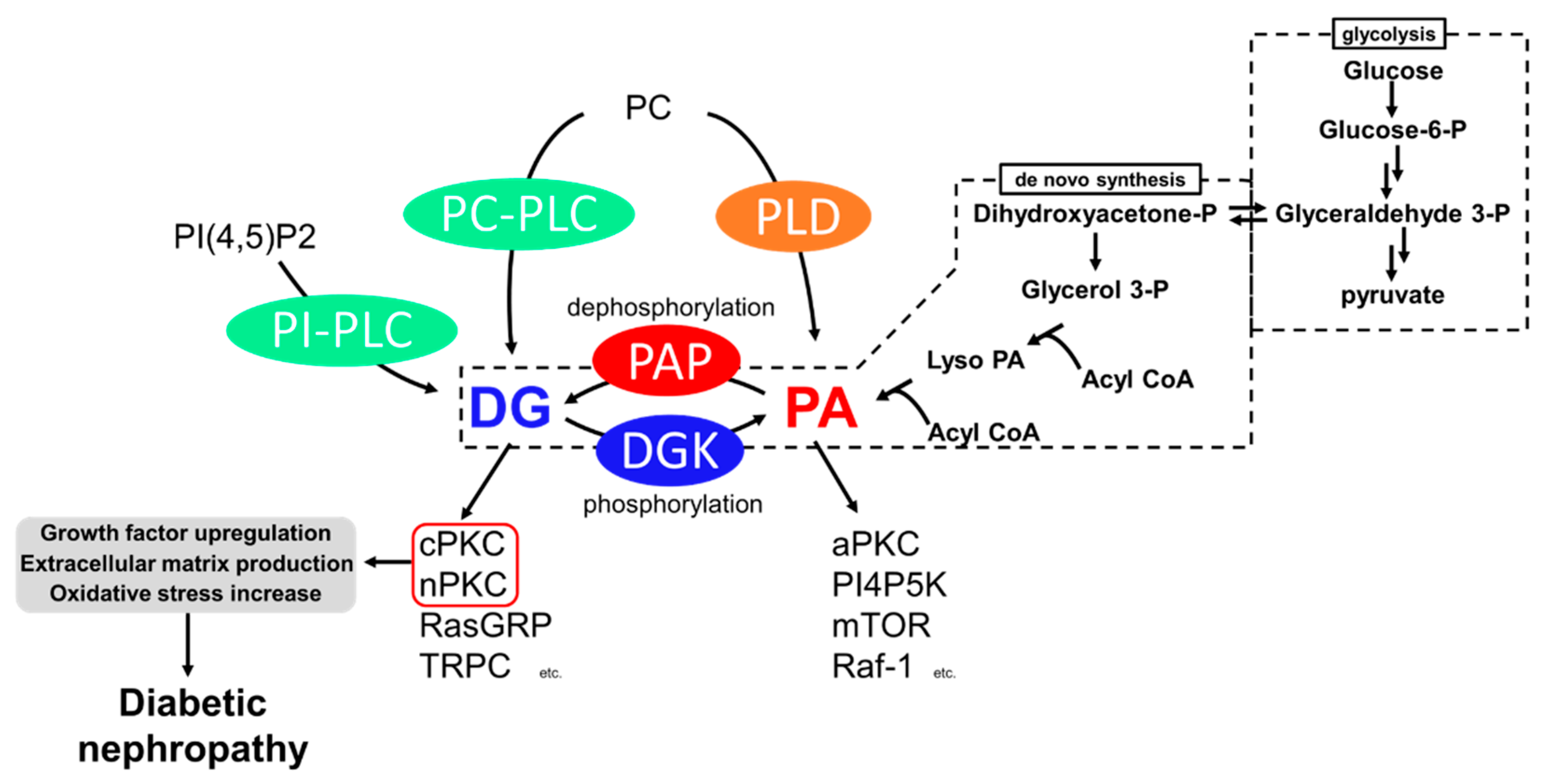
Figure 1. Generation pathways of DG and pathways leading to diabetic nephropathy. P: phosphate.
3. Diabetic Nephropathy and PKC
PKC is a family of serine/threonine kinases, and nine mammalian isozymes are categorized into three groups based on their domain structure and characteristics [23]. cPKC (PKCα, β, γ) possesses diacylglycerol (DG) binding C1 domains (C1A and C1B) and a Ca2+ binding C2 domain, and their activity depends on DG and Ca2+ [24][25]. nPKC (PKCδ, ε, η, θ) has a slightly different domain composition than cPKC and lost Ca2+ dependency [26]. The DG-responsive characteristics of cPKC and nPKC are crucial to the pathogenesis of DN. The final subfamily requires neither Ca2+ nor DG for its activity and was cloned and classified as aPKC (PKCι, ζ). It is activated by PA [27][28]. These PKCs phosphorylate many proteins, including receptors, transporters, kinases, and phosphatases; thus, PKCs play a central role in cell signaling [23]. To date, the increase in DG mass and the abnormal activation of cPKC and nPKC under hyperglycemic conditions have been observed in vivo and in vitro [29][30][31], and evidence supports the idea that the abnormal activation of cPKC and nPKC is one of the causes of the development and progression of diabetic nephropathy [32][33][34][35].
Abnormally activated PKC causes various biological changes, such as growth factor upregulation, extracellular matrix production, and oxidative stress increase (Figure 1). Among PKC isozymes, activation of PKCα, β (cPKC) and PKCε, δ (nPKC) have been observed in DN [28][34][35][36][37][38]. One of the mechanisms underlying how PKC activation causes DN is related to various growth factors [39]. The upregulation of transforming growth factor-β (TGF-β) signaling, a profibrotic cytokine, is considered a critical pathogenic and progression factor of DN by activating various signal transductions, expression of the extracellular matrix, and reactive oxygen species production [40][41]. It has been reported that diabetes-induced activation of PKCβ and PKCδ upregulates TGF-β expression and the sensitivity of the cell against TGF-β, promoting the production of the extracellular matrix [42][43]. In addition to TGF-β, connective tissue growth factor (CTGF) and vascular endothelial growth factor (VEGF) are also recognized as key factors in DN progression [37]. CTGF is known to promote extracellular matrix production, leading to fibrosis [44]. VEGF is known as an angiogenic factor promoting vascular endothelial cell proliferation and pathological angiogenesis [45]. Several studies have reported that activation of PKCα, β, and ε upregulates VEGF expression [46][47], and PKCβ upregulates CTGF expression [34]. Therefore, PKC inhibition can normalize these growth factor expressions and signaling and ameliorate DN. Moreover, PKCα and β are associated with reactive oxygen species (ROS) production by activation of NADPH oxidase, causing renal injury [34][48].
To develop drugs targeting abnormal PKC activation, a PKCβ-specific inhibitor, ruboxistaurin, was developed and showed a renoprotective effect in several animal studies [49][50]. However, in human clinical trials, although ruboxistaurin showed some renoprotective effect in diabetes patients, statistical significance was not observed compared to placebo patients [35]. Furthermore, since DG–PKC pathway activation is a common event in other diabetic microvascular complications, ruboxistaurin underwent phase 3 clinical trials for diabetic retinopathy. Nevertheless, the trial failed due to the insignificance of its effect on retinopathy compared to the placebo group [51]. Taken together, the upregulation of the DG–PKC pathway in diabetes is one of the key events in the pathogenesis and progression of DN. However, since several independent PKC isoforms are involved in complex and multifactorial pathways, developing a drug targeting PKC is not simple. For example, PKCα deficiency prevents albuminuria by reducing VEGF. However, glomerular hypertrophy is not inhibited due to no change in TGF-β expression [33]. In contrast, PKCβ deficiency prevents glomerular hypertrophy by reducing TGF-β but not albuminuria in mice [34][52]. Therefore, inhibiting a single PKC might be insufficient to prevent DN. One can overcome this by developing a pan-inhibitor for cPKC and nPKC, or a combination of several specific inhibitors, though issues with side effects may emerge.
4. Diabetic Nephropathy and DGK
DG kinase (DGK) is a family of enzymes that converts DG into PA by phosphorylation [53]. As illustrated in Figure 1, DGK is a central regulator in various signaling pathways related to DG and PA [54]. Since DGKs consume DG, DGK is recognized as an indirect inhibitor of cPKC and nPKC. So far, 10 subtypes of mammalian DGKs have been discovered and divided into Type I~V based on their structural characteristics [54][55][56][57]. All mammalian DGKs have several C1 domains homologous to the one of PKCs at the nitrogen ends of catalytic domains (CD). Type I DGKs (DGKα, β, γ) have a recoverin homology (RVH) domain and two EF-hand motifs, which bind to Ca2+ at the nitrogen end of the two C1 domains [58]. Type II DGKs (DGKδ, η, κ) are characterized by a split CD, with a pleckstrin homology domain (PH domain) at the N terminus. Among Type II DGKs, DGKδ and η have a sterile α motif domain (SAM domain) at the C terminus [59][60][61]. DGKε has a simple structural feature composed of two C1 domains and a CD and is the only Type III DGK subtype [62]. Type IV DGKs (DGKζ, ι) possess ankyrin repeats at the C terminus, and residue between the C1 domains and the CD have homologs to the myristoylated alanine-rich C-kinase substrate phosphorylation site [63][64][65]. Type V DGKs (DGKθ) is the only subtype that has three C1 domains, and a PH domain overlapping the Ras-associating domain (RA domain) is localized between the C1 domains and the CD [66]. In addition, splicing variants to alter their domain composition has been reported in several subtypes.
Vitamin E is a hydrophobic antioxidant agent, and α-tocopherol is the most active vitamin E compound that humans preferentially absorb [67]. The main function of α-tocopherol is its antioxidant effect, preventing lipid peroxidation in the cell membrane [68]. In 1997, it was reported that α-tocopherol ameliorates the symptoms of DN by activating DGK and inhibiting glomerulus PKC in diabetic rats, suggesting the possibility of DGK as a therapeutic target for DN [69][70]. However, the subtype of DGK involved in the amelioration was unclear. In 2005, it was revealed that a Type I DGK, DGKα, is sensitive to α-tocopherol among the DGKs expressed in the murine glomeruli (DGKα, γ, δ, ε, and ζ) [71], i.e., in the cultured cell, α-tocopherol induced translocation of GFP tagged DGKα to the plasma membrane, which is the hallmark of DGK activation, while the other DGKs did not [72][73]. Although the primary function of α-tocopherol as a vitamin is its antioxidant effect, several studies have reported that the antioxidant effect of α-tocopherol is not related to PKC inhibition [74][75]. Indeed, it was revealed that the antioxidant effect of α-tocopherol is not necessary to activate DGKα [73]. There is a report showing that the antioxidant effect of α-tocopherol plays a protective role against the loss of activity of DGKα under the high glucose condition [76]. The detailed mechanisms of DGKα activation by α-tocopherol are discussed in a later section.
References
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, Regional and Country-Level Diabetes Prevalence Estimates for 2021 and Projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119.
- Sarwar, N.; Gao, P.; Kondapally Seshasai, S.R.; Gobin, R.; Kaptoge, S.; Di Angelantonio, E.; Ingelsson, E.; Lawlor, D.A.; Selvin, E.; Stampfer, M.; et al. Diabetes Mellitus, Fasting Blood Glucose Concentration, and Risk of Vascular Disease: A Collaborative Meta-Analysis of 102 Prospective Studies. Lancet 2010, 375, 2215–2222.
- Carey, I.M.; Critchley, J.A.; Dewilde, S.; Harris, T.; Hosking, F.J.; Cook, D.G. Risk of Infection in Type 1 and Type 2 Diabetes Compared with the General Population: A Matched Cohort Study. Diabetes Care 2018, 41, 513–521.
- Hovind, P.; Tarnow, L.; Rossing, P.; Jensen, B.R.; Graae, M.; Torp, I.; Binder, C.; Parving, H.H. Predictors for the Development of Microalbuminuria and Macroalbuminuria in Patients with Type 1 Diabetes: Inception Cohort Study. Br. Med. J. 2004, 328, 1105–1108.
- Samsu, N. Diabetic Nephropathy: Challenges in Pathogenesis, Diagnosis, and Treatment. Biomed. Res. Int. 2021, 2021, 1497449.
- Secrist, J.P.; Karnitz, L.; Abraham, R.T. T-Cell Antigen Receptor Ligation Induces Tyrosine Phosphorylation of Phospholipase C-Γ1. J. Biol. Chem. 1991, 266, 12135–12139.
- Rhee, S.G.; Bae, Y.S. Regulation of Phosphoinositide-Specific Phospholipase C Isozymes. J. Biol. Chem. 1997, 272, 15045–15048.
- Monick, M.M.; Carter, A.B.; Gudmundsson, G.; Mallampalli, R.; Powers, L.S.; Hunninghake, G.W. A Phosphatidylcholine-Specific Phospholipase C Regulates Activation of P42/44 Mitogen-Activated Protein Kinases in Lipopolysaccharide-Stimulated Human Alveolar Macrophages. J. Immunol. 1999, 162, 3005–3012.
- Carman, G.M.; Han, G.S. Phosphatidic Acid Phosphatase, a Key Enzyme in the Regulation of Lipid Synthesis. J. Biol. Chem. 2009, 284, 2593–2597.
- Carman, G.M.; Han, G.-S. Roles of Phosphatidate Phosphatase Enzymes in Lipid Metabolism. Trends Biochem. Sci. 2006, 31, 694–699.
- Exton, J.H. Phospholipase D: Enzymology, Mechanisms of Regulation, and Function. Physiol. Rev. 1997, 77, 303–320.
- Craven, P.A.; Davidson, C.M.; DeRubertis, F.R. Increase in Diacylglycerol Mass in Isolated Glomeruli by Glucose from de Novo Synthesis of Glycerolipids. Diabetes 1990, 39, 667–674.
- Thorens, B.; Mueckler, M. Glucose Transporters in the 21st Century. Am. J. Physiol. Endocrinol. Metab. 2010, 298, 141–145.
- Jiang, X.; Yang, F.; Brailoiu, E.; Jakubowski, H.; Dun, N.J.; Schafer, A.I.; Yang, X.; Durante, W.; Wang, H. Differential Regulation of Homocysteine Transport in Vascular Endothelial and Smooth Muscle Cells. Diabetes 1993, 42, 80–89.
- Nishizuka, Y. Intracellular Signaling by Hydrolysis of Phospholipids and Activation of Protein Kinase, C. Science 1992, 258, 607–614.
- Newton, A.C. Regulation of Protein Kinase, C. Curr. Opin. Cell Biol. 1997, 9, 161–167.
- Ebinu, J.O.; Bottorff, D.A.; Chan, E.Y.W.; Stang, S.L.; Dunn, R.J.; Stone, J.C. RasGRP, a Ras Guanyl Nucleotide- Releasing Protein with Calcium- and Diacylglycerol-Binding Motifs. Science 1998, 280, 1082–1086.
- Lucas, P.; Ukhanov, K.; Leinders-Zufall, T.; Zufall, F. A Diacylglycerol-Gated Cation Channel in Vomeronasal Neuron Dendrites Is Impaired in TRPC2 Mutant Mice: Mechanism of Pheromone Transduction. Neuron 2003, 40, 551–561.
- Limatola, C.; Schaap, D.; Moolenaar, W.H.; van Blitterswijk, W.J. Phosphatidic Acid Activation of Protein Kinase C-Zeta Overexpressed in COS Cells: Comparison with Other Protein Kinase C Isotypes and Other Acidic Lipids. Biochem. J. 1994, 304, 1001–1008.
- Jones, D.R.; Sanjuan, M.A.; Mérida, I. Type Iα Phosphatidylinositol 4-Phosphate 5-Kinase Is a Putative Target for Increased Intracellular Phosphatidic Acid. FEBS Lett. 2000, 476, 160–165.
- Ghosh, S.; Strum, J.C.; Sciorra, V.A.; Daniel, L.; Bell, R.M. Raf-1 Kinase Possesses Distinct Binding Domains for Phosphatidylserine and Phosphatidic Acid: Phosphatidic Acid Regulates the Translocation of Raf-1 in 12-O-Tetradecanoylphorbol-13-Acetate-Stimulated Madin-Darby Canine Kidney Cells. J. Biol. Chem. 1996, 271, 8472–8480.
- Fang, Y.; Vilella-Bach, M.; Bachmann, R.; Flanigan, A.; Chen, J. Phosphatidic Acid-Mediated Mitogenic Activation of MTOR Signaling. Science 2001, 294, 1942–1945.
- Newton, A.C. Protein Kinase C: Perfectly Balanced. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 208–230.
- Parker, P.J.; Coussens, L.; Totty, N.; Rhee, L.; Young, S.; Chen, E.; Stabel, S.; Waterfield, M.D.; Ullrich, A. The Complete Primary Structure of Protein Kinase C—The Major Phorbol Ester Receptor. Science 1986, 233, 853–859.
- Knopf, J.L.; Lee, M.H.; Sultzman, L.A.; Kriz, R.W.; Loomis, C.R.; Hewick, R.M.; Bell, R.M. Cloning and Expression of Multiple Protein Kinase C CDNAs. Cell 1986, 46, 491–502.
- Ohno, S.; Akita, Y.; Konno, Y.; Imajoh, S.; Suzuki, K. A Novel Phorbol Ester Receptor/Protein Kinase, NPKC, Distantly Related to the Protein Kinase C Family. Cell 1988, 53, 731–741.
- Ono, Y.; Fujii, T.; Ogita, K.; Kikkawa, U.; Igarashi, K.; Nishizuka, Y. The Structure, Expression, and Properties of Additional Members of the Protein Kinase C Family. J. Biol. Chem. 1988, 263, 6927–6932.
- Nakanishi, H.; Exton, J.H. Purification and Characterization of the ζ Isoform of Protein Kinase C from Bovine Kidney. J. Biol. Chem. 1992, 267, 16347–16354.
- Craven, P.A.; DeRubertis, F. Protein Kinase C Is Activated in Glomeruli from Streptozotocin Diabetic Rats. Possible Mediation by Glucose. J. Clin. Investig. 1989, 83, 1667–1675.
- Inoguchi, T.; Battan, R.; Handler, E.; Sportsman, J.R.; Heath, W.; King, G.L. Preferential Elevation of Protein Kinase C Isoform Beta II and Diacylglycerol Levels in the Aorta and Heart of Diabetic Rats: Differential Reversibility to Glycemic Control by Islet Cell Transplantation. Proc. Natl. Acad. Sci. USA 1992, 89, 11059–11063.
- Ayo, S.H.; Radnik, R.; Garoni, J.A.; Troyer, D.A.; Kreisberg, J.I. High Glucose Increases Diacylglycerol Mass and Activates Protein Kinase C in Mesangial Cell Cultures. Am. J. Physiol. Physiol. 1991, 261, F571–F577.
- Koya, D.; King, G.L. Protein Kinase C Activation and the Development of Diabetic Complications. Diabetes 1998, 47, 859–866.
- Menne, J.; Park, J.K.; Boehne, M.; Elger, M.; Lindschau, C.; Kirsch, T.; Meier, M.; Gueler, F.; Fiebeler, A.; Bahlmann, F.H.; et al. Diminished Loss of Proteoglycans and Lack of Albuminuria in Protein Kinase C-α-Deficient Diabetic Mice. Diabetes 2004, 53, 2101–2109.
- Ohshiro, Y.; Ma, R.C.; Yasuda, Y.; Hiraoka-Yamamoto, J.; Clermont, A.C.; Isshiki, K.; Yagi, K.; Arikawa, E.; Kern, T.S.; King, G.L. Reduction of Diabetes-Induced Oxidative Stress, Fibrotic Cytokine Expression, and Renal Dysfunction in Protein Kinase Cβ-Null Mice. Diabetes 2006, 55, 3112–3120.
- Tuttle, K.R.; Bakris, G.L.; Toto, R.D.; McGill, J.B.; Hu, K.; Anderson, P.W. The Effect of Ruboxistaurin on Nephropathy in Type 2 Diabetes. Diabetes Care 2005, 28, 2686–2690.
- Shiba, T.; Inoguchi, T.; Sportsman, J.R.; Heath, W.F.; Bursell, S.; King, G.L. Correlation of Diacylglycerol Level and Protein Kinase C Activity in Rat Retina to Retinal Circulation. Am. J. Physiol. 1993, 265, E783–E793.
- Derubertis, F.R.; Craven, P.A. Activation of Protein Kinase C in Glomerular Cells in Diabetes: Mechanisms and Potential Links to the Pathogenesis of Diabetic Glomerulopathy. Diabetes 1994, 43, 1–8.
- Babazono, T.; Kapor-Drezgic, J.; Dlugosz, J.A.; Whiteside, C. Altered Expression and Subcellular Localization of Diacylglycerol- Sensitive Protein Kinase C Isoforms in Diabetic Rat Glomerular Cells. Diabetes 1998, 47, 668–676.
- Chiarelli, F.; Gaspari, S.; Marcovecchio, M.L. Role of Growth Factors in Diabetic Kidney Disease. Horm. Metab. Res. 2009, 41, 585–593.
- Reeves, W.B.; Andreoli, T.E. Transforming Growth Factor β Contributes to Progressive Diabetic Nephropathy. Proc. Natl. Acad. Sci. USA 2000, 97, 7667–7669.
- Arora, M.K.; Singh, U.K. Molecular Mechanisms in the Pathogenesis of Diabetic Nephropathy: An Update. Vascul. Pharmacol. 2013, 58, 259–271.
- Koya, D.; Jirousek, M.R.; Lin, Y.W.; Ishii, H.; Kuboki, K.; King, G.L. Characterization of Protein Kinase C β Isoform Activation on the Gene Expression of Transforming Growth Factor-β, Extracellular Matrix Components, and Prostanoids in the Glomeruli of Diabetic Rats. J. Clin. Investig. 1997, 100, 115–126.
- Hayashida, T.; Schnaper, H.W. High Ambient Glucose Enhances Sensitivity to TGF-Β1 via Extracellular Signal-Regulated Kinase and Protein Kinase Cδ Activities in Human Mesangial Cells. J. Am. Soc. Nephrol. 2004, 15, 2032–2041.
- Riser, B.L.; Denichilo, M.; Cortes, P.; Baker, C.; Grondin, J.M.; Yee, J.; Narins, R.G. Regulation of Connective Tissue Growth Factor Activity in Cultured Rat Mesangial Cells and Its Expression in Experimental Diabetic Glomerulosclerosis. J. Am. Soc. Nephrol. 2000, 11, 25–38.
- Khamaisi, M.; Schrijvers, B.F.; De Vriese, A.S.; Raz, I.; Flyvbjerg, A. The Emerging Role of VEGF in Diabetic Kidney Disease. Nephrol. Dial. Transplant. 2003, 18, 1427–1430.
- Hoshi, S.; Tomari, S.; Hoshi, S.; Nomoto, K.I.; Kuromitsu, J.; Nagata, M. High Glucose Induced VEGF Expression via PKC and ERK in Glomerular Podocytes. Biochem. Biophys. Res. Commun. 2002, 290, 177–184.
- Xia, L.; Wang, H.; Munk, S.; Frecker, H.; Goldberg, H.J.; Fantus, I.G.; Whiteside, C.I. Reactive Oxygen Species, PKC-A1, and PKC-ζ Mediate High-Glucose-Induced Vascular Endothelial Growth Factor Expression in Mesangial Cells. Am. J. Physiol. Endocrinol. Metab. 2007, 293, 1280–1288.
- Thallas-Bonke, V.; Thorpe, S.R.; Coughlan, M.T.; Fukami, K.; Yap, F.Y.T.; Sourris, K.C.; Penfold, S.A.; Bach, L.A.; Cooper, M.E.; Forbes, J.M. Inhibition of NADPH Oxidase Prevents Advanced Glycation End Product-Mediated Damage in Diabetic Nephropathy through a Protein Kinase C-α-Dependent Pathway. Diabetes 2008, 57, 460–469.
- Ishii, H.; Jirousek, M.R.; Koya, D.; Takagi, C.; Xia, P.; Clermont, A.; Bursell, S.E.; Kern, T.S.; Ballas, L.M.; Heath, W.F.; et al. Amelioration of Vascular Dysfunctions in Diabetic Rats by an Oral PKC β Inhibitor. Science 1996, 272, 728–731.
- Koya, D.; Haneda, M.; Nakagawa, H.; Isshiki, K.; Sato, H.; Maeda, S.; Sugimoto, T.; Yasuda, H.; Kashiwagi, A.; Ways, D.K.; et al. Amelioration of Accelerated Diabetic Mesangial Expansion by Treatment with a PKC β Inhibitor in Diabetic Db/Db Mice, a Rodent Model for Type 2 Diabetes. FASEB J. 2000, 14, 439–447.
- Sheetz, M.J.; Aiello, L.P.; Davis, M.D.; Danis, R.; Bek, T.; Cunha-Vaz, J.; Shahri, N.; Berg, P.H. The Effect of the Oral PKC β Inhibitor Ruboxistaurin on Vision Loss in Two Phase 3 Studies. Investig. Ophthalmol. Vis. Sci. 2013, 54, 1750–1757.
- Meier, M.; Park, J.K.; Overheu, D.; Kirsch, T.; Lindschau, C.; Gueler, F.; Leitges, M.; Menne, J.; Haller, H. Deletion of Protein Kinase C-β Isoform in Vivo Reduces Renal Hypertrophy but Not Albuminuria in the Streptozotocin-Induced Diabetic Mouse Model. Diabetes 2007, 56, 346–354.
- Kanoh, H.; Yamada, K.; Sakane, F. Diacylglycerol Kinase: A Key Modulator of Signal Transduction? Trends Biochem. Sci. 1990, 15, 47–50.
- Sakane, F.; Imai, S.I.; Kai, M.; Yasuda, S.; Kanoh, H. Diacylglycerol Kinases: Why so Many of Them? Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2007, 1771, 793–806.
- Topham, M.K.; Prescott, S.M. Mammalian diacylglycerol kinases, a Family of Lipid Kinases with Signaling Functions. J. Biol. Chem. 1999, 274, 11447–11450.
- Van Blitterswijk, W.J.; Houssa, B. Properties and Functions of Diacylglycerol Kinases. Cell. Signal. 2000, 12, 595–605.
- Kanoh, H.; Yamada, K.; Sakane, F. Diacylglycerol Kinases: Emerging Downstream Regulators in Cell Signaling Systems. J. Biochem. 2002, 131, 629–633.
- Sakane, F.; Yamada, K.; Kanoh, H.; Yokoyama, C.; Tanabe, T. Porcine Diacylglycerol Kinase Sequence Has Zinc Finger and E-F Hand Motifs. Nature 1990, 344, 345–348.
- Klauck, T.M.; Xu, X.; Mousseau, B.; Jaken, S. Cloning and Characterization of a Glucocorticoid-Induced Diacylglycerol Kinase. J. Biol. Chem. 1996, 271, 19781–19788.
- Letwin, K.; Yee, S.P.; Pawson, T. Novel Protein-Tyrosine Kinase CDNAs Related to Fps/Fes and Eph Cloned Using Anti-Phosphotyrosine Antibody. Oncogene 1988, 3, 621–627.
- Imai, S.; Kai, M.; Yasuda, S.; Kanoh, H.; Sakane, F. Identification and Characterization of a Novel Human Type II Diacylglycerol Kinase, DGK Kappa. J. Biol. Chem. 2005, 280, 39870–39881.
- Tang, W.; Bunting, M.; Zimmerman, G.A.; McIntyre, T.M.; Prescott, S.M. Molecular Cloning of a Novel Human Diacylglycerol Kinase Highly Selective for Arachidonate-Containing Substrates. J. Biol. Chem. 1996, 271, 10237–10241.
- Bunting, M.; Tang, W.; Zimmerman, G.A.; McIntyre, T.M.; Prescott, S.M. Molecular Cloning and Characterization of a Novel Human Diacylglycerol Kinase Zeta. J. Biol. Chem. 1996, 271, 10230–10236.
- Goto, K.; Kondo, H. A 104-KDa Diacylglycerol Kinase Containing Ankyrin-like Repeats Localizes in the Cell Nucleus. Proc. Natl. Acad. Sci. USA 1996, 93, 11196–11201.
- Ding, L.; Traer, E.; McIntyre, T.M.; Zimmerman, G.A.; Prescott, S.M. The Cloning and Characterization of a Novel Human Diacylglycerol Kinase, DGKiota. J. Biol. Chem. 1998, 273, 32746–32752.
- Houssa, B.; Schaap, D.; Van Der Wal, J.; Goto, K.; Kondo, H.; Yamakawa, A.; Shibata, M.; Takenawa, T.; Van Blitterswijk, W.J. Cloning of a Novel Human Diacylglycerol Kinase (DGKθ) Containing Three Cysteine-Rich Domains, a Proline-Rich Region, and a Pleckstrin Homology Domain with an Overlapping Ras-Associating Domain. J. Biol. Chem. 1997, 272, 10422–10428.
- Rigotti, A. Absorption, Transport, and Tissue Delivery of Vitamin E. Mol. Asp. Med. 2007, 28, 423–436.
- Traber, M.G.; Atkinson, J. Vitamin E, Antioxidant and Nothing More. Free Radic. Biol. Med. 2007, 43, 4–15.
- Koya, D.; Lee, I.K.; Ishii, H.; Kanoh, H.; King, G.L. Prevention of Glomerular Dysfunction in Diabetic Rats by Treatment with D-Alpha-Tocopherol. J. Am. Soc. Nephrol. 1997, 8, 426–435.
- Tada, H.; Ishii, H.; Isogai, S. Protective Effect of D-α-Tocopherol on the Function of Human Mesangial Cells Exposed to High Glucose Concentrations. Metabolism 1997, 46, 779–784.
- Kakehi, T.; Yagi, K.; Saito, N.; Shirai, Y. Effects of Vitamin E and Its Derivatives on Diabetic Nephropathy in Rats and Identification of Diacylglycerol Kinase Subtype Involved in the Improvement of Diabetic Nephropathy. Funct. Foods Health Dis. 2017, 7, 816–832.
- Shirai, Y.; Segawa, S.; Kuriyama, M.; Goto, K.; Sakai, N.; Naoaki, S. Subtype-Specific Translocation of Diacylglycerol Kinase α and γ and Its Correlation with Protein Kinase C. J. Biol. Chem. 2000, 275, 24760–24766.
- Fukunaga-Takenaka, R.; Shirai, Y.; Yagi, K.; Adachi, N.; Sakai, N.; Merino, E.; Merida, I.; Saito, N. Importance of Chroman Ring and Tyrosine Phosphorylation in the Subtype-Specific Translocation and Activation of Diacylglycerol Kinase α by D-α-Tocopherol. Genes Cells 2005, 10, 311–319.
- Tasinato, A.; Boscoboinik, D.; Bartoli, G.M.; Maroni, P.; Azzi, A. D-α-Tocopherol Inhibition of Vascular Smooth Muscle Cell Proliferation Occurs at Physiological Concentrations, Correlates with Protein Kinase C Inhibition, and Is Independent of Its Antioxidant Properties. Proc. Natl. Acad. Sci. USA 1995, 92, 12190–12194.
- Pryor, W.A.; Cornicelli, J.A.; Devall, L.J.; Tait, B.; Trivedi, B.K.; Witiak, D.T.; Wu, M. A Rapid Screening Test To Determine the Antioxidant Potencies of Natural and Synthetic Antioxidants. J. Org. Chem. 1993, 58, 3521–3532.
- Atsumi, H.; Kitada, M.; Kanasaki, K.; Koya, D. Reversal of Redox-Dependent Inhibition of Diacylglycerol Kinase by Antioxidants in Mesangial Cells Exposed to High Glucose. Mol. Med. Rep. 2011, 4, 923–927.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
998
Revisions:
2 times
(View History)
Update Date:
27 Oct 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No