 +1 credit
+1 credit
Video Upload Options
Haemophilia, or hemophilia (from grc αἷμα (Script error: No such module "Ancient Greek".) 'blood', and φιλία (Script error: No such module "Ancient Greek".) 'love of'), is a mostly inherited genetic disorder that impairs the body's ability to make blood clots, a process needed to stop bleeding. This results in people bleeding for a longer time after an injury, easy bruising, and an increased risk of bleeding inside joints or the brain. Those with a mild case of the disease may have symptoms only after an accident or during surgery. Bleeding into a joint can result in permanent damage while bleeding in the brain can result in long term headaches, seizures, or a decreased level of consciousness. There are two main types of haemophilia: haemophilia A, which occurs due to low amounts of clotting factor VIII, and haemophilia B, which occurs due to low levels of clotting factor IX. They are typically inherited from one's parents through an X chromosome carrying a nonfunctional gene. Rarely a new mutation may occur during early development or haemophilia may develop later in life due to antibodies forming against a clotting factor. Other types include haemophilia C, which occurs due to low levels of factor XI, and parahaemophilia, which occurs due to low levels of factor V. Acquired haemophilia is associated with cancers, autoimmune disorders, and pregnancy. Diagnosis is by testing the blood for its ability to clot and its levels of clotting factors. Prevention may occur by removing an egg, fertilizing it, and testing the embryo before transferring it to the uterus, which is connected with ethical problems. Human embryos in research can be regarded as the technical object/process. Missing blood clotting factors are replaced to treat haemophilia. This may be done on a regular basis or during bleeding episodes. Replacement may take place at home or in hospital. The clotting factors are made either from human blood or by recombinant methods. Up to 20% of people develop antibodies to the clotting factors which makes treatment more difficult. The medication desmopressin may be used in those with mild haemophilia A. Studies of gene therapy are in early human trials. Haemophilia A affects about 1 in 5,000–10,000, while haemophilia B affects about 1 in 40,000, males at birth. As haemophilia A and B are both X-linked recessive disorders, females are rarely severely affected. Some females with a nonfunctional gene on one of the X chromosomes may be mildly symptomatic. Haemophilia C occurs equally in both sexes and is mostly found in Ashkenazi Jews. In the 1800s haemophilia B was common within the royal families of Europe. The difference between haemophilia A and B was determined in 1952.
1. Signs and Symptoms
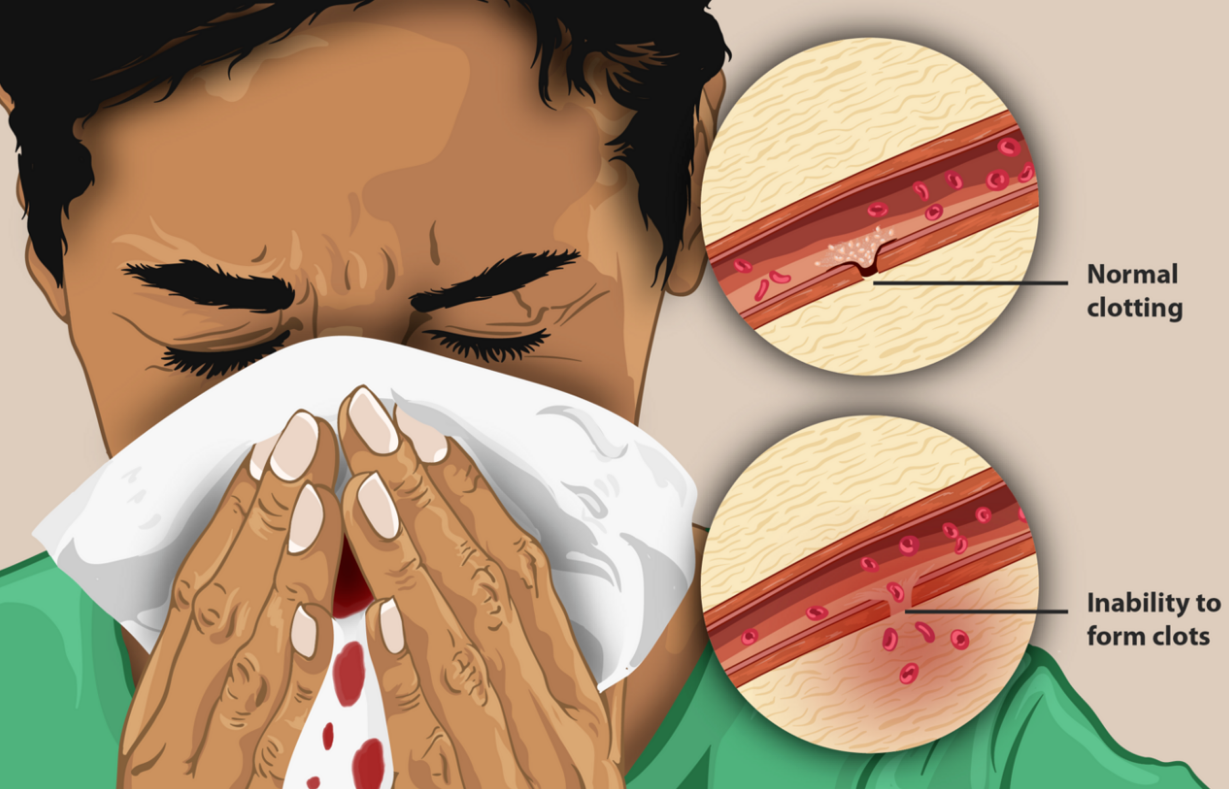
Characteristic symptoms vary with severity. In general symptoms are internal or external bleeding episodes, which are called "bleeds".[1][2] People with more severe haemophilia experience more severe and more frequent bleeds, while people with mild haemophilia usually experience more minor symptoms except after surgery or serious trauma. In cases of moderate haemophilia symptoms are variable which manifest along a spectrum between severe and mild forms.[3]
In both haemophilia A and B, there is spontaneous bleeding but a normal bleeding time, normal prothrombin time, normal thrombin time, but prolonged partial thromboplastin time. Internal bleeding is common in people with severe haemophilia and some individuals with moderate haemophilia. The most characteristic type of internal bleed is a joint bleed where blood enters into the joint spaces.[4] This is most common with severe haemophiliacs and can occur spontaneously (without evident trauma). If not treated promptly, joint bleeds can lead to permanent joint damage and disfigurement.[4] Bleeding into soft tissues such as muscles and subcutaneous tissues is less severe but can lead to damage and requires treatment.
Children with mild to moderate haemophilia may not have any signs or symptoms at birth, especially if they do not undergo circumcision. Their first symptoms are often frequent and large bruises and haematomas from frequent bumps and falls as they learn to walk. Swelling and bruising from bleeding in the joints, soft tissue, and muscles may also occur. Children with mild haemophilia may not have noticeable symptoms for many years. Often, the first sign in very mild haemophiliacs is heavy bleeding from a dental procedure, an accident, or surgery. Females who are carriers usually have enough clotting factors from their one normal gene to prevent serious bleeding problems, though some may present as mild haemophiliacs.
1.1. Complications
Severe complications are much more common in cases of severe and moderate haemophilia. Complications may arise from the disease itself or from its treatment:[5]
- Deep internal bleeding, e.g. deep-muscle bleeding, leading to swelling, numbness or pain of a limb.
- Joint damage from haemarthrosis (haemophilic arthropathy), potentially with severe pain, disfigurement, and even destruction of the joint and development of debilitating arthritis.
- Transfusion transmitted infection from blood transfusions that are given as treatment.
- Adverse reactions to clotting factor treatment, including the development of an immune inhibitor which renders factor replacement less effective.
- Intracranial haemorrhage is a serious medical emergency caused by the buildup of pressure inside the skull. It can cause disorientation, nausea, loss of consciousness, brain damage, and death.
Haemophilic arthropathy is characterized by chronic proliferative synovitis and cartilage destruction.[6] If an intra-articular bleed is not drained early, it may cause apoptosis of chondrocytes and affect the synthesis of proteoglycans. The hypertrophied and fragile synovial lining while attempting to eliminate excessive blood may be more likely to easily rebleed, leading to a vicious cycle of hemarthrosis-synovitis-hemarthrosis. In addition, iron deposition in the synovium may induce an inflammatory response activating the immune system and stimulating angiogenesis, resulting in cartilage and bone destruction.[7]
2. Genetics
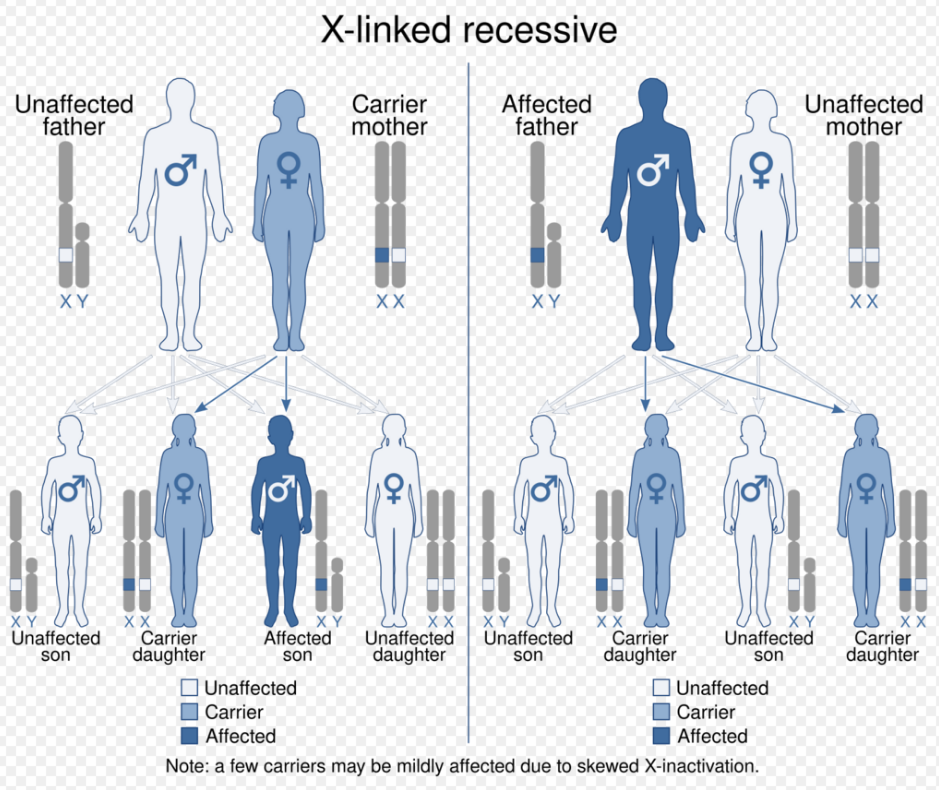
Typically, females possess two X-chromosomes, and males have one X and one Y-chromosome. Since the mutations causing the disease are X-linked recessive, a female carrying the defect on one of her X-chromosomes may not be affected by it, as the equivalent dominant allele on her other chromosome should express itself to produce the necessary clotting factors, due to X inactivation. Therefore, heterozygous females are just carriers of this genetic disposition. However, the Y-chromosome in the male has no gene for factors VIII or IX. If the genes responsible for production of factor VIII or factor IX present on a male's X-chromosome are deficient there is no equivalent on the Y-chromosome to cancel it out, so the deficient gene is not masked and the disorder will develop.[8]
Since a male receives his single X-chromosome from his mother, the son of a healthy female silently carrying the deficient gene will have a 50% chance of inheriting that gene from her and with it the disease; and if his mother is affected with haemophilia, he will have a 100% chance of being a haemophiliac. In contrast, for a female to inherit the disease, she must receive two deficient X-chromosomes, one from her mother and the other from her father (who must therefore be a haemophiliac himself). Hence, haemophilia is expressed far more commonly among males than females, while females, who must have two deficient X-chromosomes in order to have haemophilia, are far more likely to be silent carriers, survive childhood and to submit each of her genetic children to an at least 50% risk of receiving the deficient gene. However, it is possible for female carriers to become mild haemophiliacs due to lyonisation (inactivation) of the X-chromosomes. Haemophiliac daughters are more common than they once were, as improved treatments for the disease have allowed more haemophiliac males to survive to adulthood and become parents. Adult females may experience menorrhagia (heavy periods) due to the bleeding tendency. The pattern of inheritance is criss-cross type. This type of pattern is also seen in colour blindness.
A mother who is a carrier has a 50% chance of passing the faulty X-chromosome to her daughter, while an affected father will always pass on the affected gene to his daughters. A son cannot inherit the defective gene from his father. This is a recessive trait and can be passed on if cases are more severe with carrier. Genetic testing and genetic counselling is recommended for families with haemophilia. Prenatal testing, such as amniocentesis, is available to pregnant women who may be carriers of the condition.[9]
As with all genetic disorders, it is also possible for a human to acquire it spontaneously through mutation, rather than inheriting it, because of a new mutation in one of their parents' gametes. Spontaneous mutations account for about 33% of all cases of haemophilia A.[10] About 30% of cases of haemophilia B are the result of a spontaneous gene mutation.
If a female gives birth to a haemophiliac son, either the female is a carrier for the blood disorder or the haemophilia was the result of a spontaneous mutation.[11] Until modern direct DNA testing, however, it was impossible to determine if a female with only healthy children was a carrier or not.[12]
If a male is has the disease and has children with a female who is not a carrier, his daughters will be carriers of haemophilia. His sons, however, will not be affected with the disease. The disease is X-linked and the father cannot pass haemophilia through the Y-chromosome. Males with the disorder are then no more likely to pass on the gene to their children than carrier females, though all daughters they sire will be carriers and all sons they father will not have haemophilia (unless the mother is a carrier).
2.1. Severity
There are numerous different mutations which cause each type of haemophilia. Due to differences in changes to the genes involved, people with haemophilia often have some level of active clotting factor. Individuals with less than 1% active factor are classified as having severe haemophilia, those with 1–5% active factor have moderate haemophilia, and those with mild haemophilia have between 5% and 40% of normal levels of active clotting factor.[4]
3. Diagnosis
Haemophilia can be diagnosed before, during or after birth if there is a family history of the condition. Several options are available to parents. If there is no family history of haemophilia, it is usually only diagnosed when a child begins to walk or crawl. They may experience joint bleeds or easy bruising.[13]
Mild haemophilia may only be discovered later, usually after an injury or a dental or surgical procedure.
3.1. Before Pregnancy
Genetic testing and counselling are available to help determine the risk of passing the condition onto a child.[13] This may involve testing a sample of tissue or blood to look for signs of the genetic mutation that causes haemophilia.[13]
3.2. During Pregnancy
A pregnant woman with a history of haemophilia in her family can test for the haemophilia gene. Such tests include:
- chorionic villus sampling (CVS): a small sample of the placenta is removed from the womb and tested for the haemophilia gene, usually during weeks 11–14 of pregnancy[14]
- amniocentesis: a sample of amniotic fluid is taken for testing, usually during weeks 15–20 of pregnancy[15]
There is a small risk of these procedures causing problems such as miscarriage or premature labour, so the woman may discuss this with the doctor in charge of her care.[13]
3.3. After Birth
If haemophilia is suspected after a child has been born, a blood test can usually confirm the diagnosis. Blood from the umbilical cord can be tested at birth if there's a family history of haemophilia. A blood test will also be able to identify whether a child has haemophilia A or B, and how severe it is.[13]
3.4. Classification
There are several types of haemophilia: haemophilia A, haemophilia B, haemophilia C, parahaemophilia, acquired haemophilia A, and acquired haemophilia B.[16][17][18][19]
Haemophilia A is a recessive X-linked genetic disorder resulting in a deficiency of functional clotting Factor VIII.[17] Haemophilia B is also a recessive X-linked genetic disorder involving a lack of functional clotting Factor IX.[20] Haemophilia C is an autosomal genetic disorder involving a lack of functional clotting Factor XI. Haemophilia C is not completely recessive, as heterozygous individuals also show increased bleeding.[18]
The type of haemophilia known as parahaemophilia is a mild and rare form and is due to a deficiency in factor V. This type can be inherited or acquired.[21]
A non-genetic form of haemophilia is caused by autoantibodies against factor VIII and so is known as acquired haemophilia A.[22] It is a rare but potentially life-threatening bleeding disorder caused by the development of autoantibodies (inhibitors) directed against plasma coagulation factors.[23] Acquired haemophilia can be associated with cancers, autoimmune disorders and following childbirth.[24]
4. Management
There is no long-term cure. Treatment and prevention of bleeding episodes is done primarily by replacing the missing blood clotting factors.[25]
4.1. Clotting Factors
Clotting factors are usually not needed in mild haemophilia.[26] In moderate haemophilia clotting factors are typically only needed when bleeding occurs or to prevent bleeding with certain events.[26] In severe haemophilia preventive use is often recommended two or three times a week and may continue for life.[26] Rapid treatment of bleeding episodes decreases damage to the body.[26]
Factor VIII is used in haemophilia A and factor IX in haemophilia B. Factor replacement can be either isolated from human blood serum, recombinant, or a combination of the two. Some people develop antibodies (inhibitors) against the replacement factors given to them, so the amount of the factor has to be increased or non-human replacement products must be given, such as porcine factor VIII.[27]
If a person becomes refractory to replacement coagulation factor as a result of high levels of circulating inhibitors, this may be partially overcome with recombinant human factor VIII.[28]
In early 2008, the US Food and Drug Administration (FDA) approved an anti-haemophilic drug completely free of albumin, which made it the first anti-haemophilic drug in the US to use an entirely synthetic purification process.[29] Since 1993 recombinant factor products (which are typically cultured in Chinese hamster ovary (CHO) tissue culture cells and involve little, if any human plasma products) have been available and have been widely used in wealthier western countries. While recombinant clotting factor products offer higher purity and safety, they are, like concentrate, extremely expensive, and not generally available in the developing world. In many cases, factor products of any sort are difficult to obtain in developing countries.
Clotting factors are either given preventively or on-demand. Preventive use involves the infusion of clotting factor on a regular schedule in order to keep clotting levels sufficiently high to prevent spontaneous bleeding episodes. On-demand (or episodic) treatment involves treating bleeding episodes once they arise. In 2007, a trial comparing on-demand treatment of boys (< 30 months) with haemophilia A with prophylactic treatment (infusions of 25 IU/kg body weight of Factor VIII every other day) in respect to its effect on the prevention of joint-diseases. When the boys reached 6 years of age, 93% of those in the prophylaxis group and 55% of those in the episodic-therapy group had a normal index joint-structure on MRI.[30] Preventative treatment, however, resulted in average costs of United States dollar 300,000 per year. The author of an editorial published in the same issue of the NEJM supports the idea that prophylactic treatment not only is more effective than on demand treatment but also suggests that starting after the first serious joint-related haemorrhage may be more cost effective than waiting until the fixed age to begin.[31] Most haemophiliacs in third world countries have limited or no access to commercial blood clotting factor products.[32]
4.2. Other
Desmopressin (DDAVP) may be used in those with mild haemophilia A.[26] Tranexamic acid or epsilon aminocaproic acid may be given along with clotting factors to prevent breakdown of clots.[26]
Pain medicines, steroids, and physical therapy may be used to reduce pain and swelling in an affected joint.[26] In those with severe hemophilia A already receiving FVIII, emicizumab may provide some benefit.[33] Different treatments are used to help those with an acquired form of hemophilia in addition to the normal clotting factors. Often the most effective treatment is corticosteroids which remove the auto-antibodies in half of people. As a secondary route of treatment, cyclophosphamide and cyclosporine are used and are proven effective for those who did not respond to the steroid treatments. In rare cases a third route or treatment is used, high doses of intravenous immunoglobulin or immunosorbent that works to help control bleeding instead of battling the auto-antibodies.[34]
4.3. Contraindications
Anticoagulants such as heparin and warfarin are contraindicated for people with haemophilia as these can aggravate clotting difficulties. Also contraindicated are those drugs which have "blood thinning" side effects. For instance, medicines which contain aspirin, ibuprofen, or naproxen sodium should not be taken because they are well known to have the side effect of prolonged bleeding.[35]
Also contraindicated are activities with a high likelihood of trauma, such as motorcycling and skateboarding. Popular sports with very high rates of physical contact and injuries such as American football, hockey, boxing, wrestling, and rugby should be avoided by people with haemophilia.[35][36] Other active sports like soccer, baseball, and basketball also have a high rate of injuries, but have overall less contact and should be undertaken cautiously and only in consultation with a doctor.[35]
5. Prognosis
Like most aspects of the disorder, life expectancy varies with severity and adequate treatment. People with severe haemophilia who do not receive adequate, modern treatment have greatly shortened lifespans and often do not reach maturity. Prior to the 1960s when effective treatment became available, average life expectancy was only 11 years.[4] By the 1980s the life span of the average haemophiliac receiving appropriate treatment was 50–60 years.[4] Today with appropriate treatment, males with haemophilia typically have a near normal quality of life with an average lifespan approximately 10 years shorter than an unaffected male.[37]
Since the 1980s the primary leading cause of death of people with severe haemophilia has shifted from haemorrhage to HIV/AIDS acquired through treatment with contaminated blood products.[4] The second leading cause of death related to severe haemophilia complications is intracranial haemorrhage which today accounts for one third of all deaths of people with haemophilia. Two other major causes of death include hepatitis infections causing cirrhosis and obstruction of air or blood flow due to soft tissue haemorrhage.[4]
6. Epidemiology
Haemophilia is rare, with only about 1 instance in every 10,000 births (or 1 in 5,000 male births) for haemophilia A and 1 in 50,000 births for haemophilia B.[38] About 18,000 people in the United States have haemophilia. Each year in the US, about 400 babies are born with the disorder. Haemophilia usually occurs in males and less often in females.[39] It is estimated that about 2,500 Canadians have haemophilia A, and about 500 Canadians have haemophilia B.[40]
7. History
John C. Otto, 1803[41]
7.1. Scientific Discovery
The excessive bleeding was known to ancient people. The Talmud instructs that a boy must not be circumcised if he had two brothers who died due to complications arising from their circumcisions, and Maimonides says that this excluded paternal half-brothers.[42] This may have been due to a concern about hemophilia.[43] The first medical professional to describe the disease was Arab surgeon Al-Zahrawi, also known as Abulcasis. In the tenth century he described families whose males died of bleeding after only minor traumas.[44] While many other such descriptive and practical references to the disease appear throughout historical writings, scientific analysis did not begin until the start of the nineteenth century.
In 1803, John Conrad Otto, a Philadelphian physician, wrote an account about "a hemorrhagic disposition existing in certain families" in which he called the affected males "bleeders".[45] He recognised that the disorder was hereditary and that it affected mostly males and was passed down by healthy females. His paper was the second paper to describe important characteristics of an X-linked genetic disorder (the first paper being a description of colour blindness by John Dalton who studied his own family). Otto was able to trace the disease back to a woman who settled near Plymouth, New Hampshire, in 1720. The idea that affected males could pass the trait onto their unaffected daughters was not described until 1813 when John F. Hay, published an account in The New England Journal of Medicine.[46][47]
In 1924, a Finnish doctor discovered a hereditary bleeding disorder similar to haemophilia localised in Åland, southwest of Finland.[48] This bleeding disorder is called "Von Willebrand Disease".
The term "haemophilia" is derived from the term "haemorrhaphilia" which was used in a description of the condition written by Friedrich Hopff in 1828, while he was a student at the University of Zurich.[45][49] In 1937, Patek and Taylor, two doctors from Harvard, discovered anti-haemophilic globulin.[50] In 1947, Pavlosky, a doctor from Buenos Aires, found haemophilia A and haemophilia B to be separate diseases by doing a lab test. This test was done by transferring the blood of one haemophiliac to another haemophiliac. The fact that this corrected the clotting problem showed that there was more than one form of haemophilia.
7.2. European Royalty
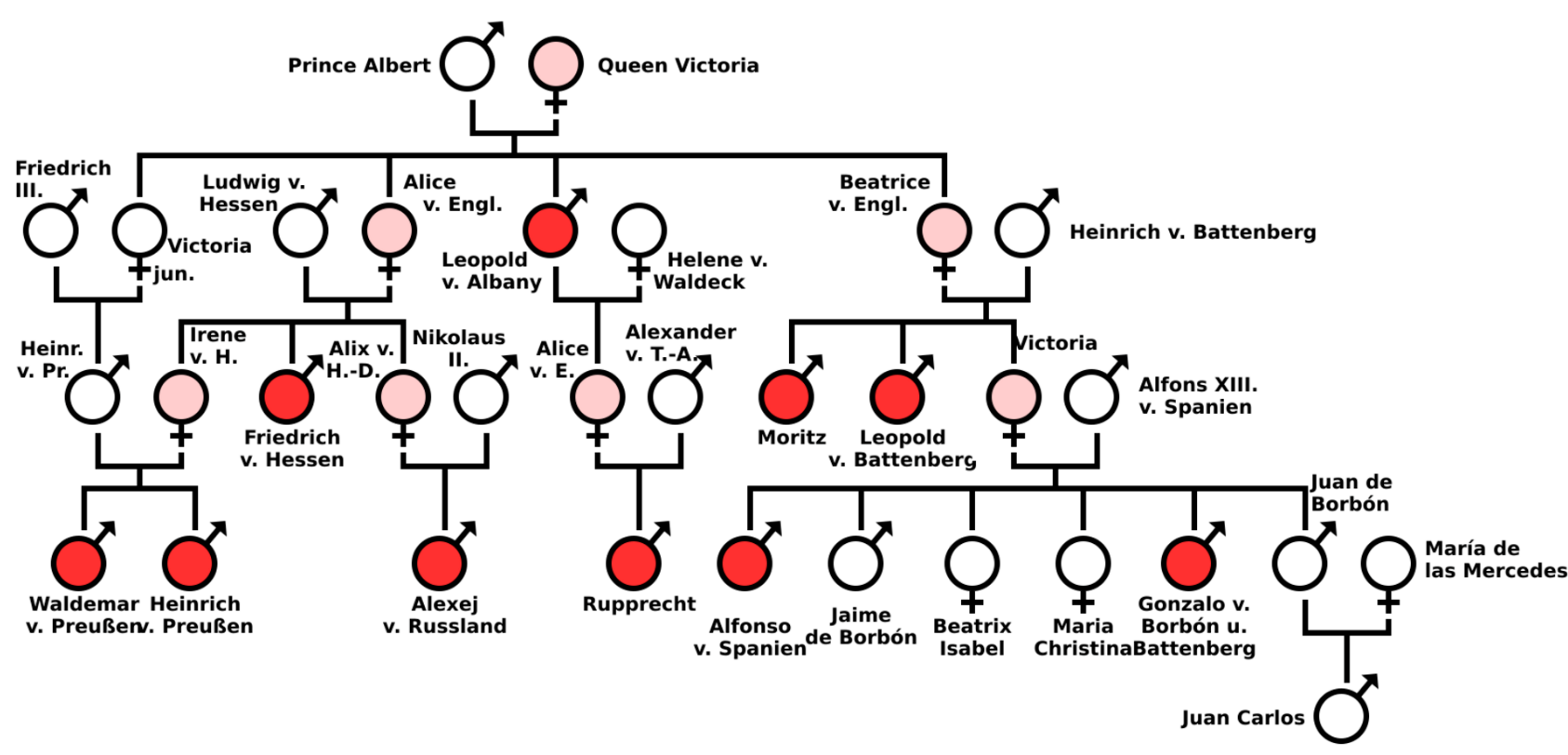
Haemophilia has featured prominently in European royalty and thus is sometimes known as 'the royal disease'. Queen Victoria passed the mutation for haemophilia B[51][52] to her son Leopold and, through two of her daughters, Alice and Beatrice, to various royals across the continent, including the royal families of Spain, Germany, and Russia. In Russia, Tsarevich Alexei, the son and heir of Tsar Nicholas II, famously had haemophilia, which he had inherited from his mother, Empress Alexandra, one of Queen Victoria's granddaughters. The haemophilia of Alexei would result in the rise to prominence of the Russian mystic Grigori Rasputin, at the imperial court.
It was claimed that Rasputin was successful at treating Tsarevich Alexei's haemophilia. At the time, a common treatment administered by professional doctors was to use aspirin, which worsened rather than lessened the problem. It is believed that, by simply advising against the medical treatment, Rasputin could bring visible and significant improvement to the condition of Tsarevich Alexei.
In Spain, Queen Victoria's youngest daughter, Princess Beatrice, had a daughter Victoria Eugenie of Battenberg, who later became Queen of Spain. Two of her sons were haemophiliacs and both died from minor car accidents. Her eldest son, Prince Alfonso of Spain, Prince of Asturias, died at the age of 31 from internal bleeding after his car hit a telephone booth.[53] Her youngest son, Infante Gonzalo, died at age 19 from abdominal bleeding following a minor car accident in which he and his sister hit a wall while avoiding a cyclist. Neither appeared injured or sought immediate medical care and Gonzalo died two days later from internal bleeding.[54]
7.3. Treatment
The method for the production of an antihaemophilic factor was discovered by Judith Graham Pool from Stanford University in 1964,[55] and approved for commercial use in 1971 in the United States under the name Cryoprecipitated AHF.[56] Together with the development of a system for transportation and storage of human plasma in 1965, this was the first time an efficient treatment for haemophilia became available.[57]
7.4. Blood Contamination

Up until late 1985 many people with haemophilia received clotting factor products that posed a risk of HIV and hepatitis C infection. The plasma used to create the products was not screened or tested, nor had most of the products been subject to any form of viral inactivation.
Tens of thousands worldwide were infected as a result of contaminated factor products including more than 10,000 people in the United States,[58] 3,500 British, 1,400 Japanese,[59] 700 Canadians,[60] 250 Irish,[61] and 115 Iraqis.[62]
Infection via the tainted factor products had mostly stopped by 1986 by which time viral inactivation methods had largely been put into place,[63] although some products were shown to still be dangerous in 1987.[64]
8. Research
8.1. Gene Therapy
In those with severe haemophilia, gene therapy may reduce symptoms to those that a person with mild or moderate haemophilia might have.[65] The best results have been found in haemophilia B.[65] In 2016 early stage human research was ongoing with a few sites recruiting participants.[65] In 2017 a gene therapy trial on nine people with haemophilia A reported that high doses did better than low doses.[66][67] It is not currently an accepted treatment for haemophilia.[26]
References
- Types of Bleeds National Hemophilia Foundation. http://www.hemophilia.org/NHFWeb/MainPgs/MainNHF.aspx?menuid=191&contentid=382&rptname=bleeding
- Key facts: what is haemophilia? The Haemophilia Society. http://www.haemophilia.org.uk/index.php?content_id=87&parent=278
- "Diagnosis and care of patients with mild haemophilia: practical recommendations for clinical management". Blood Transfusion (Blood Transfus.) 16 (6): 535–544. November 2018. doi:10.2450/2017.0150-17. PMID 29328905. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=6214819
- Hemophilia Overview eMedicine from webMD. Dimitrios P Agaliotis, MD, PhD, FACP, Robert A Zaiden, MD, Fellow, and Saduman Ozturk, PA-C. Updated: 24 November 2009. http://emedicine.medscape.com/article/210104-overview
- Hemophilia Complications Mayo Clinic Staff. 16 May 2009 http://www.mayoclinic.com/health/hemophilia/DS00218/DSECTION=complications
- Rodriguez-Merchan, E. Carlos (2010). "Musculoskeletal Complications of Hemophilia". HSS J 6 (1): 37–42. doi:10.1007/s11420-009-9140-9. PMID 19921342. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2821487
- "Pathogenesis of haemophilic synovitis: experimental studies on blood-induced joint damage". Haemophilia 13 Suppl 3: 10–3. November 2007. doi:10.1111/j.1365-2516.2007.01534.x. PMID 17822515. https://dx.doi.org/10.1111%2Fj.1365-2516.2007.01534.x
- "Hemophilia" (in en). https://www.genome.gov/genetics-glossary/hemophilia.
- Naqvi, Erum. "Hemophilia and Pregnancy - Hemophilia News Today" (in en-US). https://hemophilianewstoday.com/women-with-hemophilia__trashed/pregnancy-and-hemophilia/.
- Kumar, Parveen; Clark, Michael (2009). Kumar & Clark's Clinical Medicine (7th ed.). Saunders Elsevier. ISBN 9780702029936.
- CDC (2019-04-19). "Information for Women | Hemophilia | NCBDDD | CDC" (in en-us). https://www.cdc.gov/ncbddd/hemophilia/women.html.
- Oldenburg, Johannes; Pezeshkpoor, Behnaz; Pavlova, Anna (November 2014). "Historical review on genetic analysis in hemophilia A". Seminars in Thrombosis and Hemostasis 40 (8): 895–902. doi:10.1055/s-0034-1395161. ISSN 1098-9064. PMID 25377322. https://pubmed.ncbi.nlm.nih.gov/25377322/.
- Choices, NHS. "Haemophilia - Diagnosis - NHS Choices". http://www.nhs.uk/Conditions/Haemophilia/Pages/Diagnosis.aspx.
- "Chorionic villus sampling". National Health Service. 20 July 2018. http://www.nhs.uk/conditions/chorionic-villus-sampling/pages/introduction.aspx.
- "Amniocentesis". National Health Service. 17 April 2019. http://www.nhs.uk/conditions/amniocentesis/pages/introduction.aspx.
- "What Is Hemophilia? - NHLBI, NIH". http://www.nhlbi.nih.gov/health/health-topics/topics/hemophilia.
- "Hemophilia A: MedlinePlus Medical Encyclopedia". https://www.nlm.nih.gov/medlineplus/ency/article/000538.htm.
- Prasad Mathew, MBBS, DCH, eMedicine - Hemophilia C http://www.emedicine.com/ped/topic964.htm
- Páramo, Laura; Enciso Olivera, Leonardo Jose; Noreña, Ivan; Amaya, María A.; Santacruz, Juan C. (2019-03-05). "First Case of Acquired Hemophilia B in a Patient with HIV Infection: Case Report and Literature Review". Cureus 11 (3): e4179. doi:10.7759/cureus.4179. ISSN 2168-8184. PMID 31106079. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=6504016
- "Hemophilia B: MedlinePlus Medical Encyclopedia". https://www.nlm.nih.gov/medlineplus/ency/article/000539.htm.
- Thalji, N; Camire, RM (September 2013). "Parahemophilia: new insights into factor v deficiency.". Seminars in Thrombosis and Hemostasis 39 (6): 607–12. doi:10.1055/s-0033-1349224. PMID 23893775. https://dx.doi.org/10.1055%2Fs-0033-1349224
- Mulliez, SM; Vantilborgh, A; Devreese, KM (June 2014). "Acquired hemophilia: a case report and review of the literature.". International Journal of Laboratory Hematology 36 (3): 398–407. doi:10.1111/ijlh.12210. PMID 24750687. https://dx.doi.org/10.1111%2Fijlh.12210
- Tribuzi, Susanna, Naccarato, Alessia, Pelagalli, Lorella, et al. Acquired Hemophilia A After Hepatic Yttrium-90 Radioembolization: A Case Report. A&A Case Reports. 2017;9(12):344-345. doi:10.1213/XAA.0000000000000611.
- Franchini, M; Mannucci, PM (December 2013). "Acquired haemophilia A: a 2013 update.". Thrombosis and Haemostasis 110 (6): 1114–20. doi:10.1160/TH13-05-0363. PMID 24008306. https://dx.doi.org/10.1160%2FTH13-05-0363
- "Hemophilia Facts". August 26, 2014. https://www.cdc.gov/ncbddd/hemophilia/facts.html.
- "How Is Hemophilia Treated?". July 13, 2013. https://www.nhlbi.nih.gov/health/health-topics/topics/hemophilia/treatment.
- Mannucci, Pier Mannuccio; Franchini, Massimo (2017). "Porcine recombinant factor VIII: an additional weapon to handle anti-factor VIII antibodies". Blood Transfusion 15 (4): 365–368. doi:10.2450/2016.0030-16. ISSN 1723-2007. PMID 27483484. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=5490733
- Witmer, Char; Young, Guy (2013). "Factor VIII inhibitors in hemophilia A: rationale and latest evidence". Therapeutic Advances in Hematology 4 (1): 59–72. doi:10.1177/2040620712464509. ISSN 2040-6207. PMID 23610614. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3629762
- "FDA Gives the Go-Ahead to Wyeth for Hemophilia A Therapy and Abbott for JIA Drug" (in en-US). 2008-02-22. https://www.genengnews.com/news/fda-gives-the-go-ahead-to-wyeth-for-hemophilia-a-therapy-and-abbott-for-jia-drug/.
- "Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia". N. Engl. J. Med. 357 (6): 535–544. 2007. doi:10.1056/NEJMoa067659. PMID 17687129. https://dx.doi.org/10.1056%2FNEJMoa067659
- "Prophylactic treatment for prevention of joint disease in hemophilia—cost versus benefit". N. Engl. J. Med. 357 (6): 603–605. 2007. doi:10.1056/NEJMe078098. PMID 17687136. https://dx.doi.org/10.1056%2FNEJMe078098
- "Data Collection - WFH Annual Global Survey - World Federation of Hemophilia". https://www.wfh.org/en/data-collection.
- Research, Center for Drug Evaluation and. "Approved Drugs - FDA approves emicizumab-kxwh for hemophilia A with or without factor VIII inhibitors". https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm622564.htm.
- Boggio, Lisa N.; Green, David (2001). "Acquired Hemophilia" (in en). Reviews in Clinical and Experimental Hematology 5 (4): 389–404. doi:10.1046/j.1468-0734.2001.00049.x. ISSN 1468-0734. PMID 11844135. https://dx.doi.org/10.1046%2Fj.1468-0734.2001.00049.x
- How to Deal with Hemophilia Reviewed by: Larissa Hirsch, MD 2007. kidshealth.org by Nemours. Retrieved 23 January 2010. http://kidshealth.org/kid/health_problems/blood/hemophilia.html
- "Playing it Safe: Bleeding Disorders, Sports and Exercise ". Booklet. National Hemophilia Foundation. http://www.hemophilia.org/NHFWeb/MainPgs/MainNHF.aspx?menuid=38&contentid=27
- World Federation of Hemophilia Frequently Asked Questions. 2005 http://www.wfh.org/en/page.aspx?pid=637#Life_expectancy
- World Federation of Hemophilia Frequently Asked Questions About Hemophilia http://www.wfh.org/index.asp?lang=EN&url=2/1/1_1_1_FAQ.htm
- "U.S. National Library of Medicine". https://www.nlm.nih.gov/medlineplus/hemophilia.html.
- Canadian Hemophilia Society FAQ http://www.hemophilia.ca/en/10.php
- "Otto JC. The Medical Repository. 1803; Vol VI (1): 1-4". Genmedhist.info. http://www.genmedhist.info/articles-and-papers/.
- Mishne Torah, laws of circumcision, chapter 1 law 18. It explicitly talks about multiple boys who have the same mother, regardless of whether or not they also have the same father.
- Singer, Isidore; et al., eds. (1901–1906). "Morbidity". The Jewish Encyclopedia. New York: Funk & Wagnalls.
- "Case of the Week 175". University of Utah Medical Library. http://library.med.utah.edu/WebPath/COW/COW175.html.
- Nilsson IM (1994). "Haemophilia—then and now". Sydsvenska Medicinhistoriska Sallskapets Arsskrift 31: 33–52. PMID 11640407. http://www.ncbi.nlm.nih.gov/pubmed/11640407
- DIGITISED EARLY PAPERS AND BOOKS ON HUMAN AND MEDICAL GENETICS Genetics and Medicine Historical Network, Cardiff University. http://www.genmedhist.info/Articles%20and%20Papers/
- Hay J (July 1813). "Account of a remarkable hæmorrhagic disposition, existing in many individuals of the same family". N Engl J Med Surg 2 (3): 221–5. doi:10.1056/NEJM181307010020302. PMID 30493599. PMC 5581570. https://zenodo.org/record/1620327.
- "Haemophilia Special Issue: von Willebrand's Disease: a Report from a Meeting in the Åland Islands". Haemophilia 18. 2012. doi:10.1111/hae.2012.18.issue-s6. https://dx.doi.org/10.1111%2Fhae.2012.18.issue-s6
- "The History of hemophilia". http://www.wfh.org/2/1/1_1_3_HistoryHemophilia.htm.
- Chapter 38 Coagulation Factors V and VIII by GC White and GE Gilbert in Blood: principles and practice of hematology: 2nd edition. 2003. Eds. Robert I. Handin, Samuel E. Lux, Thomas P. Stossel. ISBN:978-0-7817-1993-3. https://books.google.com/books?id=H85dwxYTKLwC&pg=RA1-PA1195#v=onepage&q=Judah%20the%20Patriarch%20hemophilia%20Talmud
- Michael Price (8 October 2009). "Case Closed: Famous Royals Suffered From Hemophilia". ScienceNOW Daily News (AAAS). http://sciencenow.sciencemag.org/cgi/content/full/2009/1008/2.
- Evgeny I. Rogaev (8 October 2009). "Genotype Analysis Identifies the Cause of the 'Royal Disease'". Science 326 (5954): 817. doi:10.1126/science.1180660. PMID 19815722. Bibcode: 2009Sci...326..817R. https://dx.doi.org/10.1126%2Fscience.1180660
- "Tragic Drama Under the Miami Moon". http://www.biscaynetimes.com/index.php?option=com_content&id=264:tragic-drama-under-the-miami-moon&Itemid=162.
- "AUTO CRASH FATAL TO SPANISH PRINCE; Don Gonzalo, 19, Succumbs to Hemophilia After Collision in Austrian Village. INFANTA BEATRIZ DRIVING Swerved Car to Avoid Hitting Bicyclist -- Ex-King Present at Son's Bedside." (in en). http://timesmachine.nytimes.com/timesmachine/1934/08/14/93638215.html?pageNumber=7.
- Pool, Judith G.; Hershgold, Edwabd J.; Pappenhagen, Albert R. (July 1964). "High-potency Antihæmophilic Factor Concentrate prepared from Cryoglobulin Precipitate". Nature 203 (4942): 312. doi:10.1038/203312a0. ISSN 0028-0836. PMID 14201780. Bibcode: 1964Natur.203..312P. https://www.semanticscholar.org/paper/fc3f2c96768c7af5aca6561d657cba1a11eabc72.
- "Alphabetical List of Licensed Establishments Including Product Approval Dates as of 30-APR-2019". FDA. https://www.fda.gov/media/76356/download.
- Pool, Judith Graham; Shannon, Angela E. (1965-12-30). "Production of High-Potency Concentrates of Antihemophilic Globulin in a Closed-Bag System: Assay in Vitro and in Vivo". New England Journal of Medicine 273 (27): 1443–1447. doi:10.1056/NEJM196512302732701. ISSN 0028-4793. PMID 5852902. https://dx.doi.org/10.1056%2FNEJM196512302732701
- "Hemophilia: an amazing 35-year journey from the depths of HIV to the threshold of cure". Trans. Am. Clin. Climatol. Assoc. 121: 61–73; discussion 74–5. 2010. PMID 20697550. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2917149
- "Japan's Response ro the Spread of HIV/AIDS". Japan Center for International Exchange. 2004. http://www.jcie.org/researchpdfs/JapanResponseAIDS/Japansurvey.pdf.
- "Commemoration of the Tainted Blood Tragedy - Canadian Hemophilia Society". http://www.hemophilia.ca/en/commemoration-of-the-tainted-blood-tragedy/.
- "Report of the Tribunal of Inquiry into the Infection with HIV and Hepatitis C of Persons with Haemophilia and Related Matters | Department of Health". http://health.gov.ie/blog/publications/report-of-the-tribunal-of-inquiry-into-the-infection-with-hiv-and-hepatitis-c-of-persons-with-haemophilia-and-related-matters/.
- Zielbauer, Paul von (4 September 2006). "Iraqis Infected by H.I.V.-Tainted Blood Try New Tool: A Lawsuit". The New York Times. https://www.nytimes.com/2006/09/04/world/middleeast/04aids.html.
- "Haemophilia, blood products and HIV infection". Scott Med J 32 (4): 109–11. August 1987. doi:10.1177/003693308703200404. PMID 3672104. https://www.semanticscholar.org/paper/52755e70911c2af645703bcb922f0e339ade754d.
- "RCMP lay 32 charges in tainted-blood case". https://www.theglobeandmail.com/news/national/rcmp-lay-32-charges-in-tainted-blood-case/article25426655/.
- Peyvandi, F; Garagiola, I; Young, G (9 July 2016). "The past and future of haemophilia: diagnosis, treatments, and its complications.". Lancet 388 (10040): 187–97. doi:10.1016/s0140-6736(15)01123-x. PMID 26897598. https://dx.doi.org/10.1016%2Fs0140-6736%2815%2901123-x
- "AAV5-Factor VIII Gene Transfer in Severe Hemophilia A". N. Engl. J. Med. 377 (26): 2519–2530. December 2017. doi:10.1056/NEJMoa1708483. PMID 29224506. http://qmro.qmul.ac.uk/xmlui/handle/123456789/31880.
- "A Cure for Hemophilia within Reach". N. Engl. J. Med. 377 (26): 2592–2593. December 2017. doi:10.1056/NEJMe1713888. PMID 29224412. https://dx.doi.org/10.1056%2FNEJMe1713888


