Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Zeti-Azura Mohamed-Hussein | -- | 3933 | 2022-10-25 04:38:45 | | | |
| 2 | Rita Xu | Meta information modification | 3933 | 2022-10-25 07:10:28 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Abdullah-Zawawi, M.; Govender, N.; Harun, S.; Muhammad, N.A.N.; Zainal, Z.; Mohamed-Hussein, Z. Multi-Omics Approaches and Resources in the Plant Kingdom. Encyclopedia. Available online: https://encyclopedia.pub/entry/31001 (accessed on 23 June 2026).
Abdullah-Zawawi M, Govender N, Harun S, Muhammad NAN, Zainal Z, Mohamed-Hussein Z. Multi-Omics Approaches and Resources in the Plant Kingdom. Encyclopedia. Available at: https://encyclopedia.pub/entry/31001. Accessed June 23, 2026.
Abdullah-Zawawi, Muhammad-Redha, Nisha Govender, Sarahani Harun, Nor Azlan Nor Muhammad, Zamri Zainal, Zeti-Azura Mohamed-Hussein. "Multi-Omics Approaches and Resources in the Plant Kingdom" Encyclopedia, https://encyclopedia.pub/entry/31001 (accessed June 23, 2026).
Abdullah-Zawawi, M., Govender, N., Harun, S., Muhammad, N.A.N., Zainal, Z., & Mohamed-Hussein, Z. (2022, October 25). Multi-Omics Approaches and Resources in the Plant Kingdom. In Encyclopedia. https://encyclopedia.pub/entry/31001
Abdullah-Zawawi, Muhammad-Redha, et al. "Multi-Omics Approaches and Resources in the Plant Kingdom." Encyclopedia. Web. 25 October, 2022.
Copy Citation
In higher plants, the complexity of a system and the components within and among species are rapidly dissected by omics technologies. Multi-omics datasets are integrated to infer and enable a comprehensive understanding of the life processes of organisms of interest. Further, growing open-source datasets coupled with the emergence of high-performance computing and development of computational tools for biological sciences have assisted in silico functional prediction of unknown genes, proteins and metabolites, otherwise known as uncharacterized.
computational approaches
functional genomics
metabolomics
systems biology
transcriptomics
1. Introduction
The plant kingdom is comprised of photosynthetic eukaryotes, mainly green plants. The enormous variations among and within plant populations include the physical forms, reproductive mechanisms, carbon assimilation strategies (photosynthesis metabolisms), growth and development and other factors such as responses against pests and pathogens, stress environments and productivity [1]. Plants are drastically subjected to constant changes that appear invisible to the human eye, otherwise regarded as unknown.
The phenotype accounts for highly flexible differences which result from the genetics (G), environment (E), and genetics by environment interaction (GXE). The deoxyribonucleic acid (DNA) molecule is the central hereditary unit, as the genetic material is passed from one generation to the other. Composed of four different nucleotides (adenine, thymine, cytosine and guanine), DNA carries gene fragments that encode protein molecules, of which protein-encoding genes contribute to a relatively minor portion (2%) of the total genetic material (genome). The major fraction (98%) of the genome is represented by non-coding sequences, which may indirectly participate in the protein-coding gene expression mechanisms and actions. The central dogma of molecular biology maintains genetic integrity at each life cycle via replication (DNA–DNA), reverse-transcription (RNA–DNA), transcription (DNA–RNA) and translation (RNA–protein) [2]. On the other hand, gene regulatory elements (enhancers and silencers), non-coding RNAs such as microRNA (miRNA), small nuclear RNA (snRNA), small nucleolar RNA (snoRNA), long non-coding RNAs (lncRNAs), and Piwi-interacting RNA (piRNA) are explicitly reported to affect gene expression levels, DNA methylation, alternative splicing events, and epigenetics [3][4].
While the study of the entire genetic material of an organism is known as genomics, the landscape of all the elemental genes expressed (transcripts) at a given time/condition is referred as the transcriptome. Transcripts are translated into protein molecules which may undergo further modifications to form small molecules of <15,000 Da (known as metabolites). These catalogues of proteins and metabolites synthesized at a given time/condition are studied in proteomics and metabolomics, respectively. Thus, transcripts, proteins and metabolites are central components driving the complexity of a biological organism. The growing application of various omics technologies has marked a burst of scientific and technological omics-based approaches offering a wealth of plant science information. “Omics” data are either interpreted independently or integrated via multi-omics analysis to understand critical questions in plant-based research [5].
Systems biology approaches (SBA) offer a plethora of virtual modelling systems equipped with in silico designs for gene function prediction [6]. Revolutionized by high-throughput omics technologies, SBA offers a vast amount of big data generated at the molecular level [7][8]. In parallel, computational biology has gained importance alongside SBA for dissecting and further improving the biological information of the target organisms per se [9][10]. Moving forward, conventional approaches that are dependent on sequence information to predict the putative biological functions (Gene Ontology classification) of a target gene have expanded robustly to accommodate organizational level-function annotations: the structural features of a given sequence, the interaction between the gene product and the cellular entity, and the phenotypic diversity of a population. In recent years, machine learning approaches and deep learning architectures such as feature-based and artificial neural networks (convolutional neural networks (CNNs) and recurrent neural networks) have been massively deployed in plant research [11][12]. The latter was evidently highly advantageous. For example, in cis-regulatory element (CRE) prediction, the CNN, in the absence of a priori knowledge on the target location, outperforms conventional k-mer enrichment, expectation maximization and Gibbs sampling methods with a lower false positive rate [13][14][15].
2. The Omics-Platform
2.1. Genomics
The development and application of next-generation sequencing (NGS) technologies have revolutionized crop improvement strategies primarily through genome exploration and gene discovery [16][17]. Genomics study infers the function and evolutionary history of plants, and with growing NGS technologies such as Illumina, Pacific Biosciences, Beijing Genomic Institute (BGI), Twist Bioscience, 10XGenomics and Oxford Nanopore, the research output (scientific publication) has significantly increased over the last decade (2012 to 2022) (Figure 1). The NGS technologies are indeed robust tools for genome characterization (genome size and genome ploidy level) and genetic variation identification at the genome and/or population level. Genomic datasets are established by means of comprehensive methods which involves the target species’ DNA isolation, sequencing and sequence annotation using bioinformatics tools. Whole-genome sequencing (WGS) requires the entire DNA content of a single organism, while exome sequencing examines the coding DNA sequences (exons) of a genome. Another technique, namely genotype by sequencing (GBS), is a combinatorial technique that employs restriction enzymes to select single nucleotide polymorphisms (SNP) within a population. Epigenomics targets the gene-regulating components such as DNA methylation [18][19].
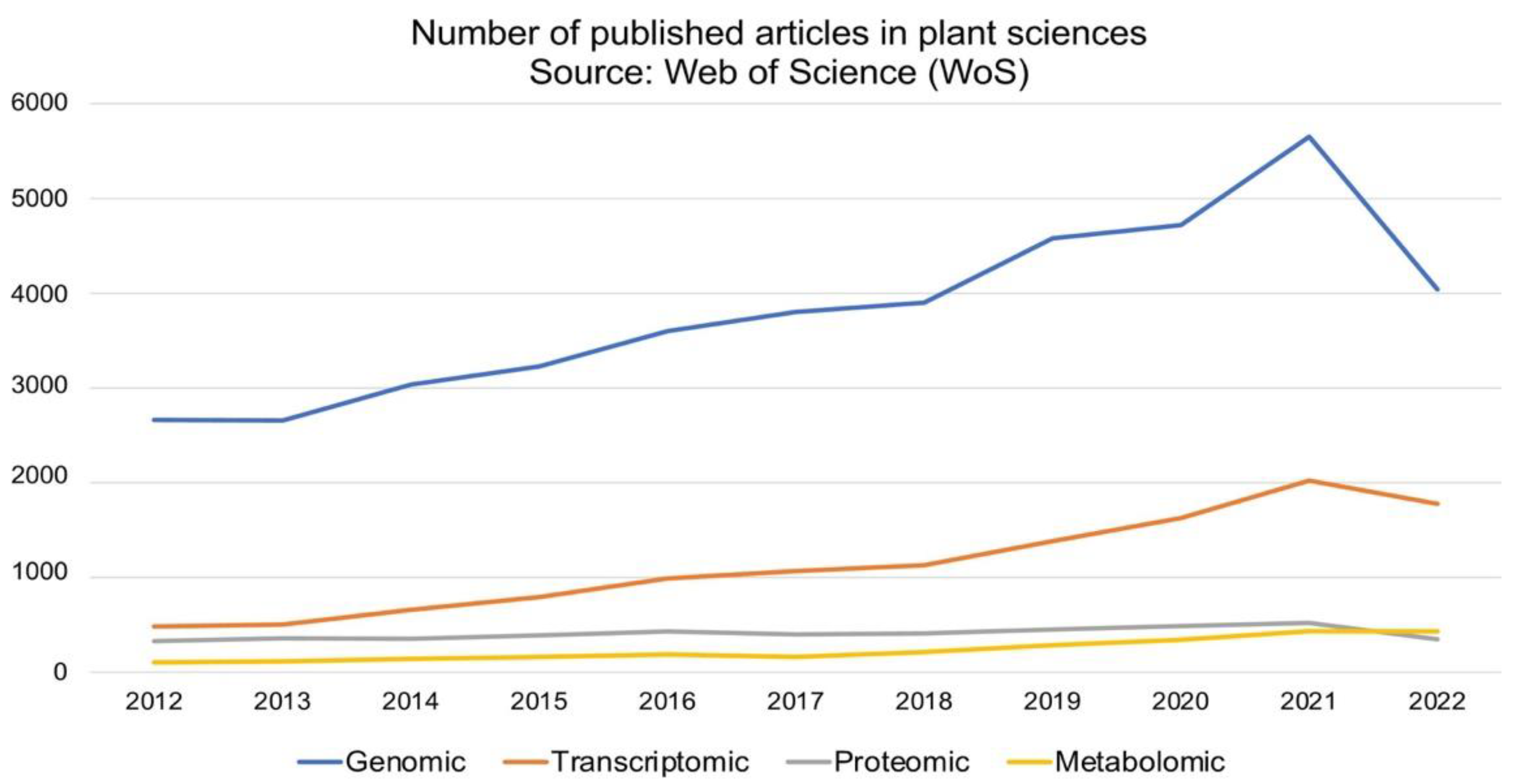
Figure 1. Scholarly omics-related articles published under the plant sciences category from 2012 to 2022. The literature search using Web of Science (https://www.webofknowledge.com) search engine was accessed on 18 September 2022 with Boolean ‘or’ and the following keywords: genomic, genome, transcriptomic, transcriptome, proteomic, proteome, metabolomic and metabolome.
The decreasing cost of genome sequencing has led to a deluge of plant genome sequences, particularly of agricultural crop sequences [20][21]. Sequencing price varies by the experimental designs and each design considers a myriad of technical features, such as number of reads, read length, methodology and technology. The most used methodologies to generate paired end reads in Illumina are Hiseq (100–250 bp) and Miseq (up to 300 bp). The latter has a low throughput and thus is highly recommended for small genomes <20 Mb. Next, PacBio emerged as a third-generation technology for complex genome sequencing of about 2.5–80 kb. The detection principle is based on the nucleotide excitation of a single molecule, and the technology is subjected to high error rates. The MinION by Oxford Nanopore sequences up to 20 Gb and comes with a low cost, portability features and a high error rate, comparatively much higher than PacBio. Another affordable NGS platform is BGISEQ, a forthcoming technology gaining a foothold in Asia. This technology generates single-end and paired-end reads of about 50–100 bp [22][23]. To date, Illumina remains the best quality read-producing technology. The quality of read profiles generated by Illumina can be evaluated in real time, and poor reads are filtered off using various user-friendly applications as follows: FastQC [24], Cutadapt [25], AdapterRemoval [26], Skewer [27], and Trimmomatic [28].
Plant genome assembly is challenged by the genome size, sequence repetitive nature and ploidy level (autoploid and alloploid). For example, a wheat genome of about 17 Gb features three independent sub-genomes [29]. The genome assembly procedure becomes easiest with the availability of a single allele per locus, although that is not the usual case in most plant genomes. In a systemic comparison between plant and vertebrate genomes using the unbiased kmer-based approach, plant genomes showed higher repeat contents [30].
Upon genome assembly, subsequent genome annotation is required to identify functional elements present along the genome sequence [31]. The genome structural annotation or gene predicting process adds biological meaning to the raw sequences and offers fundamental insights into the biology of the target species. However, the genome annotation process for high-quality genome assemblies is often challenged by the gene density and the introns abundantly present in a genome. There are three distinct computational algorithms developed for detecting the coding region; ab initio (intrinsic), evidence-based (extrinsic) and genomic sequence comparison. The ab initio gene finding prediction software includes the hidden Markov models (HMM), conditional random field, support vector machine, and neural networks. Integrating the information from both the content sensor and signal sensor [31][32][33][34][35], the content sensor classifies the DNA sequence as coding or non-coding, whilst the signal sensor identifies specific functional regions (donor or acceptor of splice site) throughout the genome [30]. Ab initio gene predictors, for instance, GenePRIMP [36], SnowyOwl [37], CodingQuarry [38], BRAKER1 [39], MAKER2 [40], MAKER-P [41] and Seqping [42], can thus be used as a pipeline to predict a reliable annotation on the newly sequenced genomes.
The evidence-based method exploits a cost-effective approach in the form of transcriptional evidence by expressed sequence tags (ESTs) or complementary DNA (cDNA) [43]. The genomic sequence comparison identifies the relativity of the content sensor to the sequence of other genomic DNA [44]. Among the notable comparative gene-finding predictors, CONTRAST [44] has a higher accuracy in both exon/gene sensitivity and specificity than any previous year predictors; N-SCAN, TWINSCAN [45] and GENSCAN [46]. The ab initio and genomic sequence comparison methods are somehow less convincing than evidence-based due to automatic prediction based on training datasets and have poor quality in algorithms that often result in errors.
Genome sequence data facilitate comparative genomic studies targeted to infer the functions of unknown genes [47][48], enable reconstruction of metabolic pathways [49][50] and advance the understanding of evolutionary relationships between and among species [51]. Genome annotation is generally performed using sequence similarity search whereby annotated genes which encode proteins are matched with known proteins available in open repositories [48][52]. To date, plant genomic information can be retrieved from public databases such as NCBI [52] and Ensembl Plants [53]. Meanwhile, PlantGDB [54], PLAZA [55], Gramene [56] and Phytozome [57].
2.1.1. Genomic-Assisted Gene Discovery for Crop Improvement
Genomics is the key enabler of the five Gs in crop improvement instruments: (i) genome assembly, (ii) germplasm characterization, (iii) gene function identification, (iv) genomic breeding and (v) gene editing [58]. Crops with established genome assemblies are research-friendly, as the ease of computational analyses is becoming highly feasible. Plant genetic resources play a fundamental role in leveraging maximum genetic gain in a breeding program. Genetic variation under the natural setting offers breeders the basis for selection and further exploitation for crop improvement. Genetic diversity of highly valuable agronomic traits such as yield, yield-related traits, and resistance against biotic and abiotic components are amongst the most widely exploited traits for further modifications [59]. Generally, mining desirable genetic variants for subsequent improvement serves as the underlying principle of crop genetic improvement. Population-level characterization of genetic variation includes the identification of deletions, insertions, transversions, copy numbers and single nucleotide polymorphisms (SNPs). A germplasm collection holds a broad genetic diversity; thus, the accurate characterization of a large-scale germplasm remains challenging. Nevertheless, advances in genotyping and phenotyping technologies have revolutionized genomic breeding (GB) approaches.
Early GB methods were developed using markers specifically associated with genes and the quantitative trait loci governing major effects of a trait per se. Such methods were extensively applied in early GB programs: marker-assisted selection (MAS), marker-assisted backcrossing (MABC) and marker-assisted recurrent selection (MARS) [60]. Later, in the quest for genetic gain and enhanced breeding efficiency, new, improved methods emerged: genome-wide association study (GWAS), expression QTL (eQTL), haplotype-based breeding, forward breeding (FB), genomic selection (GS) and speed breeding (SB) [60][61].
2.1.2. Single Cell Sequencing
A single cell is the basic structural and functional unit of living organisms. The formation and function of higher-level tissues and organs are influenced by the various genetic mechanisms along stimuli at the cellular environment. Cell heterogeneity refers to the diverse cell states formed throughout cell growth (genetic and molecular biological changes). With highly specialized structures and functions, the cells of multicellular organisms share identical genetics and sets of genetic instructions in the translation of a functional organism. Single-cell genomics offers the cell-specific landscape information regarding the organisms’ genetics, capturing the cell physiology dynamics [62].
The discovery of cell-specific transcription, tissue-specific spatial gene expression, the role of cell localization, the binding and activity of transcription factors, and the chromatin and cis-regulatory signatures of a system of interest is now feasible with growing commercial and specialized equipment systems catered toward resolving cell-specific activities. The chromatin accessibility profiling methods such as the DNase 1 hypersensitive site sequencing and assay for transposase-accessible chromatin sequencing (ATAC-seq) measure the chromatin accessibility for plant regulatory DNA across population-level species [63]. The disadvantages of these methods include a tendency to mask the cell-specific and rare events of a target tissue. Alternatively, improved high-cost systems such as the single-cell ATAC seq assays (integrated co-encapsulation or barcoding of individual cells) perform sequencing at the single-cell level [64]. In transcriptional profiling using the scRNA-seq method, the following strategies are most frequently employed: (i) fluorescence activated sorting (FACS), (ii) isolation of nuclei tagged in individual cell types (INTACT) and (iii) laser capture microdissection (LCM). Both FACS and INTACT have restricted use on selected plant species only, whereas the LCM offers a broader application range on a vast number of plant species. In general, these methods lack markers corresponding to the different differentiation states of the cell types [65].
The establishment of the Plant Cell Atlas in 2019 officially marked the trajectory of single-cell studies performed by the plant research community. Comprehensive high-resolution plant cell information (nucleic acids, proteins and metabolites) is built and shared among the scientific community [66]. Single-cell RNA sequencing (scRNA-seq) resolves cell-to-cell heterogeneity using high-throughput technologies: Drop-Seq, Chromium, Seq-well, SMART-seq 3 and iCell8 [67]. These methods offer a variety of features, which account for the following factors: (1) the target mRNA region (5′, 3′ or full length), (2) the number of cells, (3) the cell preparation technique (droplets, cell sorting and nanowells), (4) unique molecular identifiers (UMIs)—the mRNA molecule label, (5) cell size, and (6) method availability. In numerous previous studies, scRNA-seq applied on numerous tissues (Arabidopsis, rice, peanut, maize) revealed high heterogeneity, highlighting the expression signatures of cell types and development trajectories [68]. In the conventional RNA-seq method, the bulk information (average gene expression of the sample) is obtained, whereas the scRNA-seq technique consists of pools of information, each corresponding to the different types of cells present in the sample. The cell preparation is rendered as the utmost challenge to obtaining a decent result with accurate interpretations. Optimizing the protoplast isolation is vital, considering the following factors in a typical plant cell: cell density, cell wall thickness, digestion efficacy (influenced by cuticle, lignin, suberin and other deposition), enzyme type and requirement and enzyme digestion time [67][69].
2.1.3. Genome-Wide Association Study (GWAS)
Amongst these methods, GS is the most preferred tool for breeding programs, as the method does not rely on diagnostic markers entirely and the selection is made on the breeding lines evaluated according to genomic-estimated breeding values (GEBV) generated from the genomic-wide marker data sets. Genomic selection (GS) gathers the additive effects of all the genes governing the genetic variance of a given trait. With each independent gene imparting a relatively small effect, the number of genes controlling a single trait may stretch from hundreds to thousands [60][70]. Using a genome-wide marker and phenotype information, the GS method establishes the association between markers and phenotypes from an observed population. A GS analysis was first performed following Fisher’s infinite model, and soon was extended to the genomic best linear unbiased prediction (GBLUP) model. The latter accommodates GXE interactions and thus offers a more accurate prediction [61][71]. Later, the Markov chain, Monte Carlo and Bayesian modelling methods were developed to include non-additive genetic effects such as adverse environmental conditions. In the GS method, machine learning builds a training/reference population of individuals with information of interest (genotype and phenotype) to train prediction models on the test population or selection candidates. The prediction accuracy is affected by training set population size, density/number of the genome-wide markers and the heritability of the trait of interest [72].
Genomics, together with advanced-level genomic tools, open-source genome resources and powerful technologies, have accelerated crop breeding through rapid trait discovery techniques. Proposed 15 years ago, genomics-assisted breeding (GAB) has now expedited a broad range of breeding programs for resistance enhancement against diseases and tolerance improvement against abiotic factors such as submergence, salinity and drought. In rice, the “Improved Samba Mahsuri”, a GAB product, carries the Xa21, xa13, xa5 and xa38 genes governing the bacterial blight (BB) disease (causal pathogen, Xanthomonas oryzae) along with Pi-2 and Pi-54, blast disease (causal pathogen, Magnoporthe oryzae) resistance genes [73][74][75].
2.1.4. Pan-Genomics
There are about 390 thousand land plant species, and their genomes are highly complicated (highly repetitive DNA content, polyploidy and heterozygosity) and diverse (genome size varying from 60 Mb to 150 Gb). Plant genome changes arise from evolutionary forces that shaped plant speciation and evolution. Pan-genomics, a subset of plant genomic research, is highly suitable for plant species with extensive genetic diversity at the population level. Pan-genomes have been developed for important agricultural crops and model plants such as rice, Arabidopsis, barley, soybean, maize, wheat, tomato, etc. [76]. The key principles of pan-genomics include the comparison of high-quality genomes to provide insights into the collection of core and dispensable genes in a species population. Generally, a single genome or a small number of genomes do not make a good sample in pan-genome construction. Integration of many high-quality genomes is important to obtain comprehensive genetic information of the target population [77]. Genes are designated as the basic units defining a pan-genome. Pan-genome studies are most useful in understanding plants with a wide spectrum of genetic diversity and gene pools. In brief, the pan-genome strategy first establishes a target population of highly diverse individuals. A good selection of representative individuals in the population is reflected by phenotypic diversity, as determined by the phylogenetic relationship among the individuals of the population. Next, a high-quality genome assembly method for long reads is employed using automatic annotation pipelines. The construction approaches available for pan-genome analyses includes the de novo assembly (detects variant types and classifies genes into core and dispensable), iterative assembly (based on a single reference genome), and graph-based assembly strategy (utilizes graphs from a reference genome to represent the diversity and variations). Comprehensive tools and pipelines popularly employed in pan-genome analyses were exhaustively described by Li et al., 2022 [78].
2.2. Transcriptomics
A transcriptome is an atlas of RNA transcripts of a tissue, cell or defined specific condition [79]. Using the genome information, a transcriptome is “read” to obtain a comprehensive description of the genes expressed at a given time point. The mapping and quantification of the transcriptional activity are central to transcriptome studies. In the modern era, the transcriptomes are produced either by the microarray [80] or RNA-sequencing (RNA-seq) technology [81]. The latter is preferred by the plant research community due to higher precision in capturing lowly expressed RNAs and isoforms [81]. Comparatively, the RNA-seq technology detects a greater percentage of novel transcripts than the microarray [82][83]. In most transcriptome data analyses, the raw count data are subjected to differentially expressed genes (DEGs) analysis, co-expression network construction and other techniques such as alternative splicing and isoform analysis [84][85]. Both DEG and network analyses are used extensively to discover genes underpinning various biological processes such as plant defense response [86], regulation [87], water stress JAZ1 in G. arboreum [88], desiccation tolerance and drought (such as LEA) in A. thaliana seeds [89], cellulose synthase in secondary cell wall synthesis [90] and cell wall-related genes in A. thaliana [91].
In 2002, the Gene Expression Omnibus (GEO) repository was first established as an open repository for gene expression data obtained from various platforms such as microarrays, serial analysis of gene expression (SAGE) and other sequence-based data [92]. Since then, the number of open-source gene expression data repositories for various plant species and condition-specific has been on the rise: The Arabidopsis Information Resource (TAIR) [93], TRAVA [94], RiceXPro [95], Transcriptome Encyclopedia of Rice (TENOR) [96], Barley Gene Expression Database (Bex-db) [97], and Plant Stress RNA-Seq Nexus (PSRN) [98] (Table 1).
Table 1. Plant omics databases, as accessed on 24 August 2022.
| Omics Type | Database | Organism | URL | References |
|---|---|---|---|---|
| Genomics | Plant Genome Database (PlantGDB) | Plants | http://www.plantgdb.org | [54] |
| Plant Genome DataBase Japan (PGDBj) | Plants | http://pgdbj.jp/?ln=en | [99] | |
| National Center for Biotechnology Information (NCBI) | Various | https://www.ncbi.nlm.nih.gov | [52] | |
| Ensembl Plants | Plants | http://plants.ensembl.org/ | [53] | |
| Phytozome | Plants | https://phytozome.jgi.doe.gov | [57] | |
| PLAZA | Plants | https://bioinformatics.psb.ugent.be/plaza/ | [55] | |
| Plant Genome and Systems Biology (PGSB PlantsDB) | Plants | http://pgsb.helmholtz-muenchen.de/plant/plantsdb.jsp | [100] | |
| Chloroplast Genome Database (ChloroplastDB) | Plants | http://chloroplast.cbio.psu.edu/ | [101] | |
| The Solanaceae Genomics Resource (Spud DB) | Potato | http://solanaceae.plantbiology.msu.edu | [102] | |
| Melon Genome Database (Melonomics) | Melon | https://www.melonomics.net/ | [103] | |
| Maize Genetics and Genomics Database (MaizeGDB) | Maize | https://www.maizegdb.org | [104] | |
| Rice Annotation Project Database (RAP-DB) | Rice | https://rapdb.dna.affrc.go.jp | [105] | |
| Rice Genome Annotation Project (RGAP) | Rice | http://rice.plantbiology.msu.edu | [106] | |
| GrainGenes | Wheat, Barley, rye, oat | http://wheat.pw.usda.gov/GG3/ | [107] | |
| SoyBase | Soy | Soybase.org | [108] | |
| Genome Database for Rosaceae (GDR) | Rosaceae plants | https://www.rosaceae.org/ | [109] | |
| Brassica Database (BRAD) | Brassica plants | http://brassicadb.org/brad/ | [110] | |
| Transcriptomics | Gene Expression Omnibus (GEO) | Various | https://www.ncbi.nlm.nih.gov/geo/ | [92] |
| AgriSeqDB | Plants | https://expression.latrobe.edu.au/agriseqdb | [111] | |
| The Bio-Analytic Resource for Plant Biology (BAR) | Plants | http://bar.utoronto.ca | [112] | |
| and | The Arabidopsis Information Resource (TAIR) | Arabidopsis | https://www.arabidopsis.org | [93] |
| Transcriptome Variation Analysis (TRAVA) | Arabidopsis | http://travadb.org | [94] | |
| The Rice Expression Profile Database (RiceXPro) | Rice | https://ricexpro.dna.affrc.go.jp | [95] | |
| Transcriptome Encycloperdia of Rice (TENOR) | Rice | http://tenor.dna.affrc.go.jp/ | [96] | |
| Barley Gene Expression Database (Bex-db) | Barley | http://barleyflc.dna.affrc.go.jp/hvdb/ | [97] | |
| Plant Stress RNA-seq Nexus (PSRN) | Plants | http://syslab5.nchu.edu.tw | [98] | |
| Plant microRNA database (PMRD) | Plants | http://bioinformatics.cau.edu.cn/PMRD/ | [113] | |
| Interactomics | STRING | Various | https://string-db.org | [114] |
| Database of Interacting Proteins (DIP) | Various | http://dip.doe-mbi.ucla.edu | [115] | |
| Protein–Protein Interaction Database for Maize (PPIM) | Maize | http://comp-sysbio.org/ppim | [116] | |
| IntAct | Various | https://www.ebi.ac.uk/intact/ | [117] | |
| Oryza sativa Protein–Protein Interaction Network (PRIN) | Rice | http://bis.zju.edu.cn/prin/ | [118] | |
| Biomolecular Interaction Network Database (BIND) | Various | http://bind.ca | [119] | |
| The Biological General Repository for Interaction Datasets (BioGRID) | Various | https://thebiogrid.org | [120] | |
| Arabidopsis thaliana Protein Interaction Network (AtPIN) | Arabidopsis | https://atpin.bioinfoguy.net | [121] | |
| PlaPPISite | Plants | http://zzdlab.com/plappisite/index.php | [122] | |
| 3D interacting domains (3did) | Various | https://3did.irbbarcelona.org | [123] | |
| Molecular INTeraction database (MINT) | Various | http://mint.bio.uniroma2.it/mint/ | [124] | |
| ATTED-II | Plants | http://atted.jp/ | [125] | |
| CressExpress | Arabidopsis | http://cressexpress.org/ | [126] | |
| Arabidopsis Network (AraNet) | Arabidopsis | http://www.inetbio.org/aranet/ | [127] | |
| Co-expressed Biological Processes (CoP) | Plants | http://webs2.kazusa.or.jp/kagiana/cop0911/ | [128] | |
| EXPath | Plants | http://expath.itps.ncku.edu.tw/ | [129] | |
| Plant Omics Data Center (PODC) | Plants | http://bioinf.mind.meiji.ac.jp/podc/ | [130] | |
| Plant Netwrok (PlaNet) | Plants | http://aranet.mpimp-golm.mpg.de/ | [131] | |
| OryzaExpress | Rice | http://plantomics.mind.meiji.ac.jp/OryzaExpress/ | [132] | |
| PlantExpress | Rice, Arabidopsis | http://plantomics.mind.meiji.ac.jp/PlantExpress/ | [133] | |
| Rice Functionally Related Gene Expression Network Database (RiceFREND) | Rice | http://ricefrend.dna.affrc.go.jp/ | [134] | |
| Vitis vinifera Co-expression Database (VTCdb) | Grape | http://vtcdb.adelaide.edu.au/ | [135] | |
| GeneMania | Various | http://genemania.org/ | [136] | |
| A Comprehensive Systems-Biology Database (CSB.DB) | Various | http://www.csbdb.de/csbdb/home/databases.html | [137] | |
| RapaNet | Brassica | http://bioinfo.mju.ac.kr/arraynet/brassica300k/query/ | [138] | |
| Rice Expression Database (RED) | Rice | http://expression.ic4r.org | [139] | |
| PhytoNet | Various | www.gene2function.de | [140] | |
| CoNekT | Plants | https://conekt.sbs.ntu.edu.sg | [141] | |
| CoCoCoNet | Plants | https://milton.cshl.edu/CoCoCoNet | [142] |
Transcriptome data relate to the prediction of genome-scale reconstruction from previous studies: the starch biosynthesis of Manihot esculenta [143], the light and temperature acclimation in Arabidopsis thaliana [144], and the biosynthesis of biotic stress-regulated pathways (i.e., tryptophan, auxin and serotonin) in Oryza sativa [145]. High and low levels of mRNA transcription have improved the understanding of the response outcome in the genome, especially those mechanistic associations between the cellular trade-offs and epistatic gene interactions [146][147].
Transcriptome-Wide Association Studies: Prediction of Genes Governing Complex traits
Global transcriptional activity measured by the transcriptome-wide association studies (TWAS) offers a fundamental understanding of the spatiotemporal regulation of transcription events in plants [148]. Transcription causes variation, often observed as a collection of events resulting from altered coding sequences. Both mRNA and protein expression are spatial and temporal targets for selecting variations caused by the coding sequences. TWAS unravel endophenotype or variation that is predominantly caused by genetic factors. Such a feature is highly valuable for prioritizing candidate genes governing complex agronomic traits. TWAS was recently proposed as a powerful tool to predict trait-associated gene expression based on GWAS summary data [149]. TWAS, in combination with GWAS, increases the power of detection of unknown genes and offers a selection of prioritized causal genes [150][151].
References
- Parry, M.A.J.; Reynolds, M.; Salvucci, M.E.; Raines, C.; Andralojc, P.J.; Zhu, X.-G.; Price, D.G.; Condon, A.G.; Furbank, R.T. Raising yield potential of wheat. II. Increasing photosynthetic capacity and efficiency. J. Exp. Bot. 2011, 62, 453–467.
- Pramanik, D.; Shelake, R.M.; Kim, M.J.; Kim, J.-Y. CRISPR-Mediated Engineering across the Central Dogma in Plant Biology for Basic Research and Crop Improvement. Mol. Plant 2021, 14, 127–150.
- Zhang, P.; Wu, W.; Chen, Q.; Chen, M. Non-Coding RNAs and their Integrated Networks. J. Integr. Bioinform. 2019, 16, 20190027.
- Yu, Y.; Zhang, Y.; Chen, X.; Chen, Y. Plant Noncoding RNAs: Hidden Players in Development and Stress Responses. Annu. Rev. Cell Dev. Biol. 2019, 35, 407–431.
- Qian, Y.; Huang, S.-S.C. Improving plant gene regulatory network inference by integrative analysis of multi-omics and high resolution data sets. Curr. Opin. Syst. Biol. 2022, 22, 8–15.
- Herrgård, M.J.; Swainston, N.; Dobson, P.; Dunn, W.B.; Arga, K.Y.; Arvas, M.; Blüthgen, N.; Borger, S.; Costenoble, R.; Heinemann, M.; et al. A consensus yeast metabolic network reconstruction obtained from a community approach to systems biology. Nat. Biotechnol. 2008, 26, 1155–1160.
- Raikhel, N.V.; Coruzzi, G.M. Achieving the in silico plant. Systems biology and the future of plant biological research. Plant Physiol. 2003, 132, 404–409.
- Santos, F.; Boele, J.; Teusink, B. A Practical Guide to Genome-Scale Metabolic Models and Their Analysis. Methods Enzymol. 2011, 500, 509–532.
- Feist, A.M.; Henry, C.S.; Reed, J.L.; Krummenacker, M.; Joyce, A.R.; Karp, P.D.; Broadbelt, L.J.; Hatzimanikatis, V.; Palsson, B. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol. Syst. Biol. 2007, 3, 121.
- McCloskey, D.; Palsson, B.; Feist, A.M. Basic and applied uses of genome-scale metabolic network reconstructions of Escherichia coli. Mol. Syst. Biol. 2013, 9, 661.
- Mahood, E.H.; Kruse, L.H.; Moghe, G.D. Machine learning: A powerful tool for gene function prediction in plants. Appl. Plant Sci. 2020, 8, e11376.
- Mahmoud, M.; Gobet, N.; Cruz-Dávalos, D.I.; Mounier, N.; Dessimoz, C.; Sedlazeck, F.J. Structural variant calling: The long and the short of it. Genome Biol. 2019, 20, 1–14.
- Weirauch, M.T.; Cote, A.; Norel, R.; Annala, M.; Zhao, Y.; Riley, T.R.; Saez-Rodriguez, J.; Cokelaer, T.; Vedenko, A.; Talukder, S.; et al. Evaluation of methods for modeling transcription factor sequence specificity. Nat. Biotechnol. 2013, 31, 126–134.
- Vu, T.T.D.; Jung, J. Protein function prediction with gene ontology: From traditional to deep learning models. PeerJ 2021, 9, e12019.
- Metzker, M.L. Sequencing technologies—the next generation. Nat. Rev. Genet. 2010, 11, 31–46.
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145.
- Singh, S.; Rao, A.; Mishra, P.; Yadav, A.K.; Maurya, R.; Kaur, S.; Tandon, G. Bioinformatics in Next-Generation Genome Se-quencing. In Current Trends in Bioinformatics: An Insight; Wadhwa, G., Shanmughavel, P., Singh, A., Bellare, J., Eds.; Springer: Singapore, 2018; pp. 27–38.
- Kühner, S.; van Noort, V.; Betts, M.J.; Leo-Macias, A.; Batisse, C.; Rode, M.; Yamada, T.; Maier, T.; Bader, S.; Beltran-Alvarez, P.; et al. Proteome Organization in a Genome-Reduced Bacterium. Science 2009, 326, 1235–1240.
- Edwards, D.; Batley, J. Plant bioinformatics: From genome to phenome. Trends Biotechnol. 2004, 22, 232–237.
- Bolger, M.E.; Weisshaar, B.; Scholz, U.; Stein, N.; Usadel, B.; Mayer, K.F. Plant genome sequencing—Applications for crop improvement. Curr. Opin. Biotechnol. 2014, 26, 31–37.
- Cao, Y.; Fanning, S.; Proos, S.; Jordan, K.; Srikumar, S. A Review on the Applications of Next Generation Sequencing Tech-nologies as Applied to Food-Related Microbiome Studies. Front. Microbiol. 2017, 8, 1829.
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351.
- Haynes, E.; Jimenez, E.; Pardo, M.A.; Helyar, S.J. The future of NGS (Next Generation Sequencing) analysis in testing food authenticity. Food Control 2019, 101, 134–143.
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 28 July 2022).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet 2011, 17, 10–12.
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Res. Notes 2016, 9, 88.
- Jiang, H.; Lei, R.; Ding, S.-W.; Zhu, S. Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform. 2014, 15, 182.
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120.
- International Wheat Genome Sequencing Consortium (IWGSC). A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 2014, 345, 1251788.
- Jiao, W.B.; Schneeberger, K. Chromosome-level assemblies of multiple Arabidopsis genomes reveal hotspots of rear-rangements with altered evolutionary dynamics. Nat. Commun. 2020, 11, 989.
- Abril, J.F.; Castellano Hereza, S. Genome Annotation. In Encyclopedia of Bioinformatics and Computational Biology; Ranganathan, S., Gribskov, M., Schönbach, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 195–209.
- Liu, Y.; Guo, J.; Hu, G.; Zhu, H. Gene prediction in metagenomic fragments based on the SVM algorithm. BMC Bioinform. 2013, 14, S12.
- Scalzitti, N.; Jeannin-Girardon, A.; Collet, P.; Poch, O.; Thompson, J.D. A benchmark study of ab initio gene prediction methods in diverse eukaryotic organisms. BMC Genom. 2020, 21, 293.
- Wang, Z.; Chen, Y.; Li, Y. A Brief Review of Computational Gene Prediction Methods. Genom. Proteom. Bioinform. 2004, 2, 216–221.
- Huang, Y.; Chen, S.-Y.; Deng, F. Well-characterized sequence features of eukaryote genomes and implications for ab initio gene prediction. Comput. Struct. Biotechnol. J. 2016, 14, 298–303.
- Pati, A.; Ivanova, N.N.; Mikhailova, N.; Ovchinnikova, G.; Hooper, S.D.; Lykidis, A.; Kyrpides, N.C. GenePRIMP: A gene prediction improvement pipeline for prokaryotic genomes. Nat. Chem. Biol. 2010, 7, 455–457.
- Reid, I.; O’Toole, N.; Zabaneh, O.; Nourzadeh, R.; Dahdouli, M.; Abdellateef, M.; Gordon, P.M.; Soh, J.; Butler, G.; Sensen, C.W.; et al. SnowyOwl: Accurate prediction of fungal genes by using RNA-Seq and homology information to select among ab initio models. BMC Bioinform. 2014, 15, 229.
- Testa, A.C.; Hane, J.K.; Ellwood, S.R.; Oliver, R.P. CodingQuarry: Highly accurate hidden Markov model gene prediction in fungal genomes using RNA-seq transcripts. BMC Genom. 2015, 16, 170.
- Hoff, K.J.; Lange, S.; Lomsadze, A.; Borodovsky, M.; Stanke, M. BRAKER1: Unsupervised RNA-Seq-Based Genome Annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 2016, 32, 767–769.
- Holt, C.; Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 2011, 12, 491.
- Campbell, M.S.; Law, M.; Holt, C.; Stein, J.C.; Moghe, G.; Hufnagel, D.; Lei, J.; Achawanantakun, R.; Jiao, D.; Lawrence, C.J.; et al. MAKER-P: A Tool Kit for the Rapid Creation, Management, and Quality Control of Plant Genome Annotations. Plant Physiol. 2013, 164, 513–524.
- Chan, K.-L.; Rosli, R.; Tatarinova, T.V.; Hogan, M.; Firdaus-Raih, M.; Low, E.-T.L. Seqping: Gene prediction pipeline for plant genomes using self-training gene models and transcriptomic data. BMC Bioinform. 2017, 18, 1–7.
- Liang, C.; Mao, L.; Ware, D.; Stein, L. Evidence-based gene predictions in plant genomes. Genome Res. 2009, 19, 1912–1923.
- Flicek, P. Gene prediction: Compare and CONTRAST. Genome Biol. 2007, 8, 233.
- Van Baren, M.J.; Koebbe, B.C.; Brent, M.R. Using N-SCAN or TWINSCAN to predict gene structures in genomic DNA se-quences. Curr. Protoc. Bioinform. 2007, 20, 4–8.
- Richmond, T. Identification of complete gene structures in genomic DNA. Genome Biol. 2000, 1, reports222.
- Seaver, S.M.D.; Henry, C.S.; Hanson, A.D. Frontiers in metabolic reconstruction and modeling of plant genomes. J. Exp. Bot. 2012, 63, 2247–2258.
- Osterman, A.; Overbeek, R. Missing genes in metabolic pathways: A comparative genomics approach. Curr. Opin. Chem. Biol. 2003, 7, 238–251.
- De Oliveira Dal’Molin, C.G.; Nielsen, L.K. Plant genome-scale metabolic reconstruction and modelling. Curr. Opin. Biotechnol. 2013, 24, 271–277.
- Pont, C.; Wagner, S.; Kremer, A.; Orlando, L.; Plomion, C.; Salse, J. Paleogenomics: Reconstruction of plant evolutionary trajectories from modern and ancient DNA. Genome Biol. 2019, 20, 29.
- Rai, A.; Saito, K. Omics data input for metabolic modeling. Curr. Opin. Biotechnol. 2016, 37, 127–134.
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI Reference Sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005, 33, D501–D504.
- Bolser, D.; Staines, D.M.; Pritchard, E.; Kersey, P. Ensembl Plants: Integrating Tools for Visualizing, Mining, and Analyzing Plant Genomics Data. Methods Mol. Biol. 2016, 1374, 115–140.
- Dong, Q.; Schlueter, S.D.; Brendel, V. PlantGDB, plant genome database and analysis tools. Nucleic Acids Res. 2004, 32, 354D–359D.
- Proost, S.; Van Bel, M.; Vaneechoutte, D.; Van De Peer, Y.; Inzé, D.; Mueller-Roeber, B.; Vandepoele, K. PLAZA 3.0: An access point for plant comparative genomics. Nucleic Acids Res. 2015, 43, D974–D981.
- Liang, C.; Jaiswal, P.; Hebbard, C.; Avraham, S.; Buckler, E.S.; Casstevens, T.; Hurwitz, B.; McCouch, S.; Ni, J.; Pujar, A.; et al. Gramene: A growing plant comparative genomics resource. Nucleic Acids Res. 2006, 36, D947–D953.
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186.
- Varshney, R.K.; Sinha, P.; Singh, V.K.; Kumar, A.; Zhang, Q.; Bennetzen, J.L. 5Gs for crop genetic improvement. Curr. Opin. Plant Biol. 2020, 56, 190–196.
- Varshney, R.K.; Pandey, M.K.; Bohra, A.; Singh, V.K.; Thudi, M.; Saxena, R.K. Toward the sequence-based breeding in legumes in the post-genome sequencing era. Theor. Appl. Genet. 2019, 132, 797–816.
- Varshney, R.K.; Bohra, A.; Roorkiwal, M.; Barmukh, R.; Cowling, W.A.; Chitikineni, A.; Lam, H.-M.; Hickey, L.T.; Croser, J.S.; Bayer, P.E.; et al. Fast-forward breeding for a food-secure world. Trends Genet. 2021, 37, 1124–1136.
- Crossa, J.; Pérez-Rodríguez, P.; Cuevas, J.; Montesinos-López, O.; Jarquín, D.; de los Campos, G.; Burgueño, J.; González-Camacho, J.M.; Pérez-Elizalde, S.; Beyene, Y.; et al. Genomic Selection in Plant Breeding: Methods, Models, and Perspectives. Trends Plant Sci. 2017, 22, 961–975.
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330.
- Minnoye, L.; Marinov, G.K.; Krausgruber, T.; Pan, L.; Marand, A.P.; Secchia, S.; Greenleaf, W.J.; Furlong, E.E.M.; Zhao, K.; Schmitz, R.J.; et al. Chromatin accessibility profiling methods. Nat. Rev. Methods Prim. 2021, 1, 58.
- Moore, J.E.; Purcaro, M.J.; Pratt, H.E.; Epstein, C.B.; Shoresh, N.; Adrian, J.; Kawli, T.; Davis, C.A.; Dobin, A.; Kaul, R.; et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710.
- Tu, X.; Marand, A.P.; Schmitz, R.J.; Zhong, S. A combinatorial indexing strategy for low-cost epigenomic profiling of plant single cells. Plant Comm. 2022, 3, 100308.
- Rodriguez-Villalon, A.; Brady, S.M. Single cell RNA sequencing and its promise in reconstructing plant vascular cell lineages. Curr. Opin. Plant Biol. 2019, 48, 47–56.
- Rhee, S.Y.; Birnbaum, K.D.; Ehrhardt, D.W. Towards building a plant cell atlas. Trends Plant Sci. 2019, 24, 303–310.
- Denyer, T.; Timmermans, M.C. Crafting a blueprint for single-cell RNA sequencing. Trends Plant Sci. 2022, 27, 92–103.
- Giacomello, S. A new era for plant science: Spatial single-cell transcriptomics. Curr. Opin. Plant Biol. 2021, 60, 102041.
- Li, X.; Zhang, X.; Gao, S.; Cui, F.; Chen, W.; Fan, L.; Qi, Y. Single-cell RNA sequencing reveals the landscape of maize root tips and assists in identification of cell type-specific nitrate-response genes. Crop J. 2022.
- He, T.; Li, C. Harness the power of genomic selection and the potential of germplasm in crop breeding for global food security in the era with rapid climate change. Crop J. 2020, 8, 688–700.
- Cuevas, J.; Crossa, J.; Montesinos-López, O.A.; Burgueño, J.; Pérez-Rodríguez, P.; de Los Campos, G. Bayesian Genomic Pre-diction with Genotype × Environment Interaction Kernel Models. G3 Genes Genomes Genet. 2017, 7, 41–53.
- Mulesa, T.H.; Westengen, O.T. Against the grain? A historical institutional analysis of access governance of plant genetic resources for food and agriculture in Ethiopia. J. World Intellect. Prop. 2020, 23, 82–120.
- Yugander, A.; Sundaram, R.M.; Singh, K.; Ladhalakshmi, D.; Rao, L.V.S.; Madhav, M.S.; Badri, J.; Prasad, M.S.; Laha, G.S. Incorporation of the novel bacterial blight resistance gene Xa38 into the genetic background of elite rice variety Improved Samba Mahsuri. PLoS ONE 2018, 13, e0198260.
- Ratna Madhavi, K.; Rambabu, R.; Abhilash Kumar, V.; Vijay Kumar, S.; Aruna, J.; Ramesh, S.; Sundaram, R.M.; Laha, G.S.; Sheshu Madhav, M.; Prasad, M.S. Marker assisted introgression of blast (Pi-2 and Pi-54) genes into the genetic background of elite, bacterial blight resistant indica rice variety, Improved Samba Mahsuri. Euphytica 2016, 212, 331–342.
- Huang, C.; Chen, Z.; Liang, C. Oryza pan-genomics: A new foundation for future rice research and improvement. Crop J. 2021, 9, 622–632.
- Li, H.; Wang, S.; Chai, S.; Yang, Z.; Zhang, Q.; Xin, H.; Xu, Y.; Lin, S.; Chen, X.; Yao, Z.; et al. Graph-based pan-genome reveals structural and sequence variations related to agronomic traits and domestication in cucumber. Nat. Commun. 2022, 13, 682.
- Li, W.; Liu, J.; Zhang, H.; Liu, Z.; Wang, Y.; Xing, L.; He, Q.; Du, H. Plant pan-genomics: Recent advances, new challenges, and roads ahead. J. Genet. Genom. 2022.
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457.
- Govindarajan, R.; Duraiyan, J.; Kaliyappan, K.; Palanisamy, M. Microarray and its applications. J. Pharm. Bioallied. Sci. 2012, 4, S310–S312.
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63.
- Zhao, S.; Fung-Leung, W.-P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-Seq and Microarray in Transcriptome Profiling of Activated T Cells. PLoS ONE 2014, 9, e78644.
- Wilhelm, B.T.; Landry, J.-R. RNA-Seq—quantitative measurement of expression through massively parallel RNA-sequencing. Methods 2009, 48, 249–257.
- Costa-Silva, J.; Domingues, D.; Martins Lopes, F. RNA-Seq differential expression analysis: An extended review and a software tool. PLoS ONE 2017, 12, e0190152.
- Yeung, K.Y.; Medvedovic, M.; Bumgarner, R.E. From co-expression to co-regulation: How many microarray experiments do we need? Genome Biol. 2004, 5, R48.
- Ederli, L.; Dawe, A.; Pasqualini, S.; Quaglia, M.; Xiong, L.; Gehring, C. Arabidopsis flower specific defense gene expression patterns affect resistance to pathogens. Front. Plant Sci. 2015, 6, 79.
- Inoue, M.; Horimoto, K. Relationship between regulatory pattern of gene expression level and gene function. PLoS ONE 2017, 12, e0177430.
- You, Q.; Zhang, L.; Yi, X.; Zhang, K.; Yao, D.; Zhang, X.; Wang, Q.; Zhao, X.; Ling, Y.; Xu, W.; et al. Co-expression network analyses identify functional modules associated with development and stress response in Gossypium arboreum. Sci. Rep. 2016, 6, 38436.
- Costa, M.C.D.; Righetti, K.; Nijveen, H.; Yazdanpanah, F.; Ligterink, W.; Buitink, J.; Hilhorst, H.W.M. A gene co-expression network predicts functional genes controlling the re-establishment of desiccation tolerance in germinated Arabidopsis thaliana seeds. Planta 2015, 242, 435–449.
- Ruprecht, C.; Mutwil, M.; Saxe, F.; Eder, M.; Nikoloski, Z.; Persson, S. Large-Scale Co-Expression Approach to Dissect Secondary Cell Wall Formation Across Plant Species. Front. Plant Sci. 2011, 2, 23.
- Wang, S.; Yin, Y.; Ma, Q.; Tang, X.; Hao, D.; Xu, Y. Genome-scale identification of cell-wall related genes in Arabidopsis based on co-expression network analysis. BMC Plant Biol. 2012, 12, 138.
- Barrett, T.; Edgar, R. Gene Expression Omnibus: Microarray Data Storage, Submission, Retrieval, and Analysis. Methods Enzymol. 2006, 411, 352–369.
- Huala, E.; Dickerman, A.W.; Garcia-Hernandez, M.; Weems, D.; Reiser, L.; LaFond, F.; Hanley, D.; Kiphart, D.; Zhuang, M.; Huang, W.; et al. The Arabidopsis Information Resource (TAIR): A comprehensive database and web-based information re-trieval, analysis, and visualization system for a model plant. Nucleic Acids Res. 2001, 29, 102–105.
- Klepikova, A.V.; Kulakovskiy, I.V.; Kasianov, A.S.; Logacheva, M.D.; Penin, A.A. An update to database TraVA: Organ-specific cold stress response in Arabidopsis thaliana. BMC Plant Biol. 2019, 19, 29–40.
- Sato, Y.; Antonio, B.A.; Namiki, N.; Takehisa, H.; Minami, H.; Kamatsuki, K.; Sugimoto, K.; Shimizu, Y.; Hirochika, H.; Nagamura, Y. RiceXPro: A platform for monitoring gene expression in japonica rice grown under natural field conditions. Nucleic Acids Res. 2011, 39, D1141–D1148.
- Kawahara, Y.; Oono, Y.; Wakimoto, H.; Ogata, J.; Kanamori, H.; Sasaki, H.; Mori, S.; Matsumoto, T.; Itoh, T. TENOR: Database for Comprehensive mRNA-Seq Experiments in Rice. Plant Cell Physiol. 2016, 57, e7.
- Tanaka, T.; Sakai, H.; Fujii, N.; Kobayashi, F.; Nakamura, S.; Itoh, T.; Matsumoto, T.; Wu, J. bex-db: Bioinformatics workbench for comprehensive analysis of barley-expressed genes. Breed. Sci. 2013, 63, 430–434.
- Li, J.-R.; Liu, C.-C.; Sun, C.-H.; Chen, Y.-T. Plant stress RNA-seq Nexus: A stress-specific transcriptome database in plant cells. BMC Genom. 2018, 19, 966.
- Nakaya, A.; Ichihara, H.; Asamizu, E.; Shirasawa, S.; Nakamura, Y.; Tabata, S.; Hirakawa, H. Plant Genome DataBase Japan (PGDBj). Methods Mol. Biol. 2017, 1533, 45–77.
- Spannagl, M.; Nussbaumer, T.; Bader, K.C.; Martis, M.M.; Seidel, M.; Kugler, K.G.; Gundlach, H.; Mayer, K.F. PGSB PlantsDB: Updates to the database framework for comparative plant genome research. Nucleic Acids Res. 2016, 44, D1141–D1147.
- Cui, L.; Veeraraghavan, N.; Richter, A.; Wall, K.; Jansen, R.K.; Leebens-Mack, J.; Makalowska, I.; dePamphilis, C.W. Chloro-plastDB: The Chloroplast Genome Database. Nucleic Acids Res. 2006, 34, D692–D696.
- Hirsch, C.; Hamilton, J.; Childs, K.; Cepela, J.; Crisovan, E.; Vaillancourt, B.; Hirsch, C.N.; Habermann, M.; Neal, B.; Buell, C.R. Spud DB: A Resource for Mining Sequences, Genotypes, and Phenotypes to Accelerate Potato Breeding. Plant Genome 2014, 7, plantgenome2013-10.
- Ruggieri, V.; Alexiou, K.; Morata, J.; Argyris, J.; Pujol, M.; Yano, R.; Nonaka, S.; Ezura, H.; Latrasse, D.; Boualem, A.; et al. An improved assembly and annotation of the melon (Cucumis melo L.) reference genome. Sci. Rep. 2018, 8, 8088.
- Portwood, J.L., II; Woodhouse, M.R.; Cannon, E.K.; Gardiner, J.M.; Harper, L.C.; Schaeffer, M.L.; Walsh, J.R.; Sen, T.Z.; Cho, K.T.; Schott, D.A.; et al. MaizeGDB 2018: The maize multi-genome genetics and genomics database. Nucleic Acids Res. 2019, 47, D1146–D1154.
- Sakai, H.; Lee, S.S.; Tanaka, T.; Numa, H.; Kim, J.; Kawahara, Y.; Wakimoto, H.; Yang, C.-C.; Iwamoto, M.; Abe, T.; et al. Rice Annotation Project Database (RAP-DB): An Integrative and Interactive Database for Rice Genomics. Plant Cell Physiol. 2013, 54, e6.
- Kawahara, Y.; de la Bastide, M.; Hamilton, J.P.; Kanamori, H.; McCombie, W.R.; Ouyang, S.; Schwartz, D.C.; Tanaka, T.; Wu, J.; Zhou, S.; et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 2013, 6, 4.
- Yao, E.; Blake, V.C.; Cooper, L.; Wight, C.P.; Michel, S.; Cagirici, H.B.; Lazo, G.R.; Birkett, C.L.; Waring, D.J.; Jannink, J.-L.; et al. GrainGenes: A data-rich repository for small grains genetics and genomics. Database 2022, 2022, baac034.
- Brown, A.V.; Conners, S.I.; Huang, W.; Wilkey, A.P.; Grant, D.; Weeks, N.T.; Cannon, S.B.; Graham, M.A.; Nelson, R.T. A new decade and new data at SoyBase, the USDA-ARS soybean genetics and genomics database. Nucleic Acids Res. 2021, 49, D1496–D1501.
- Jung, S.; Staton, M.; Lee, T.; Blenda, A.; Svancara, R.; Abbott, A.; Main, D. GDR (Genome Database for Rosaceae): Integrated web-database for Rosaceae genomics and genetics data. Nucleic Acids Res. 2008, 36, D1034–D1040.
- Chen, H.; Wang, T.; He, X.; Cai, X.; Lin, R.; Liang, J.; Wu, J.; King, G.; Wang, X. BRAD V3.0: An upgraded Brassicaceae database. Nucleic Acids Res. 2022, 50, D1432–D1441.
- Robinson, A.J.; Tamiru, M.; Salby, R.; Bolitho, C.; Williams, A.; Huggard, S.; Fisch, E.; Unsworth, K.; Whelan, J.; Lewsey, M.G. AgriSeqDB: An online RNA-Seq database for functional studies of agriculturally relevant plant species. BMC Plant Biol. 2018, 18, 200.
- Waese, J.; Provart, N.J. The Bio-Analytic Resource for Plant Biology. In Plant Genomics Databases. Methods in Molecular Biology; Van Dijk, A., Ed.; Humana Press: New York, NJ, USA, 2017; Volume 1533, pp. 119–148.
- Zhang, Z.; Yu, J.; Li, D.; Zhang, Z.; Liu, F.; Zhou, X.; Wang, T.; Ling, Y.; Su, Z. PMRD: Plant microRNA database. Nucleic Acids Res. 2010, 38, D806–D813.
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613.
- Xenarios, I.; Rice, D.W.; Salwinski, L.; Baron, M.K.; Marcotte, E.M.; Eisenberg, D. DIP: The database of interacting proteins. Nucleic Acids Res. 2000, 28, 289–291.
- Zhu, G.; Wu, A.; Xu, X.-J.; Xiao, P.-P.; Lu, L.; Liu, J.; Cao, Y.; Chen, L.; Wu, J.; Zhao, X.-M. PPIM: A Protein-Protein Interaction Database for Maize. Plant Physiol. 2016, 170, 618–626.
- del Toro, N.; Shrivastava, A.; Ragueneau, E.; Meldal, B.; Combe, C.; Barrera, E.; Perfetto, L.; How, K.; Ratan, P.; Shirodkar, G.; et al. The IntAct database: Efficient access to fine-grained molecular interaction data. Nucleic Acids Res. 2022, 50, D648–D653.
- Gu, H.; Zhu, P.; Jiao, Y.; Meng, Y.; Chen, M. PRIN: A predicted rice interactome network. BMC Bioinform. 2011, 12, 161.
- Bader, G.; Donaldson, I.; Wolting, C.; Ouellette, B.F.F.; Pawson, T.; Hogue, C.W.V. BIND--The Biomolecular Interaction Network Database. Nucleic Acids Res. 2003, 31, 248–250.
- Chatr-Aryamontri, A.; Breitkreutz, B.-J.; Oughtred, R.; Boucher, L.; Heinicke, S.; Chen, D.; Stark, C.; Breitkreutz, A.; Kolas, N.; O’Donnell, L.; et al. The BioGRID interaction database: 2015 update. Nucleic Acids Res. 2014, 43, D470–D478.
- Brandão, M.M.; Dantas, L.L.; Silva-Filho, M.C. AtPIN: Arabidopsis thaliana Protein Interaction Network. BMC Bioinform. 2009, 10, 454.
- Yang, X.; Yang, S.; Qi, H.; Wang, T.; Li, H.; Zhang, Z. PlaPPISite: A comprehensive resource for plant protein-protein interaction sites. BMC Plant Biol. 2020, 20, 61.
- Stein, A.; Russell, R.B.; Aloy, P. 3did: Interacting protein domains of known three-dimensional structure. Nucleic Acids Res. 2005, 33, D413–D417.
- Chatr-Aryamontri, A.; Ceol, A.; Palazzi, L.M.; Nardelli, G.; Schneider, M.V.; Castagnoli, L.; Cesareni, G. MINT: The Molecular INTeraction database. Nucleic Acids Res. 2007, 35, D572–D574.
- Aoki, Y.; Okamura, Y.; Tadaka, S.; Kinoshita, K.; Obayashi, T. ATTED-II in 2016: A Plant Coexpression Database Towards Lineage-Specific Coexpression. Plant Cell Physiol. 2016, 57, e5.
- Srinivasasainagendra, V.; Page, G.P.; Mehta, T.; Coulibaly, I.; Loraine, A.E. CressExpress: A Tool for Large-Scale Mining of Expression Data from Arabidopsis. Plant Physiol. 2008, 147, 1004–1016.
- Lee, T.; Yang, S.; Kim, E.; Ko, Y.; Hwang, S.; Shin, J.; Shim, J.E.; Shim, H.; Kim, H.; Kim, C.; et al. AraNet v2: An improved database of co-functional gene networks for the study of Arabidopsis thaliana and 27 other nonmodel plant species. Nucleic Acids Res. 2015, 43, D996–D1002.
- Ogata, Y.; Suzuki, H.; Sakurai, N.; Shibata, D. CoP: A database for characterizing co-expressed gene modules with biological information in plants. Bioinformatics 2010, 26, 1267–1268.
- Chien, C.-H.; Chow, C.-N.; Wu, N.-Y.; Chiang-Hsieh, Y.-F.; Hou, P.-F.; Chang, W.-C. EXPath: A database of comparative expression analysis inferring metabolic pathways for plants. BMC Genom. 2015, 16, S6.
- Ohyanagi, H.; Takano, T.; Terashima, S.; Kobayashi, M.; Kanno, M.; Morimoto, K.; Kanegae, H.; Sasaki, Y.; Saito, M.; Asano, S.; et al. Plant Omics Data Center: An integrated web repository for interspecies gene expression networks with NLP-based cu-ration. Plant Cell Physiol. 2015, 56, e9.
- Mutwil, M.; Klie, S.; Tohge, T.; Giorgi, F.; Wilkins, O.; Campbell, M.; Fernie, A.R.; Usadel, B.; Nikoloski, Z.; Persson, S. PlaNet: Combined Sequence and Expression Comparisons across Plant Networks Derived from Seven Species. Plant Cell 2011, 23, 895–910.
- Hamada, K.; Hongo, K.; Suwabe, K.; Shimizu, A.; Nagayama, T.; Abe, R.; Kikuchi, S.; Yamamoto, N.; Fujii, T.; Yokoyama, K.; et al. OryzaExpress: An Integrated Database of Gene Expression Networks and Omics Annotations in Rice. Plant Cell Physiol. 2011, 52, 220–229.
- Kudo, T.; Terashima, S.; Takaki, Y.; Tomita, K.; Saito, M.; Kanno, M.; Yokoyama, K.; Yano, K. PlantExpress: A Database Inte-grating OryzaExpress and ArthaExpress for Single-species and Cross-species Gene Expression Network Analyses with Mi-croarray-Based Transcriptome Data. Plant Cell Physiol. 2017, 58, e1.
- Sato, Y.; Namiki, N.; Takehisa, H.; Kamatsuki, K.; Minami, H.; Ikawa, H.; Ohyanagi, H.; Sugimoto, K.; Itoh, J.-I.; Antonio, B.A.; et al. RiceFREND: A platform for retrieving coexpressed gene networks in rice. Nucleic Acids Res. 2013, 41, D1214–D1221.
- Wong, D.C.; Sweetman, C.; Drew, D.P.; Ford, C.M. VTCdb: A gene co-expression database for the crop species Vitis vinifera (grapevine). BMC Genom. 2013, 14, 882.
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220.
- Steinhauser, D.; Usadel, B.; Luedemann, A.; Thimm, O.; Kopka, J. CSB.DB: A comprehensive systems-biology database. Bioinformatics 2004, 20, 3647–3651.
- Kim, J.; Jun, K.M.; Kim, J.S.; Chae, S.; Pahk, Y.-M.; Lee, T.-H.; Sohn, S.-I.; Lee, S.I.; Lim, M.-H.; Kim, C.-K.; et al. RapaNet: A Web Tool for the Co-Expression Analysis of Brassica rapa Genes. Evol. Bioinform. 2017, 13.
- Xia, L.; Zou, D.; Sang, J.; Xu, X.; Yin, H.; Li, M.; Wu, S.; Hu, S.; Hao, L.; Zhang, Z. Rice Expression Database (RED): An integrated RNA-Seq-derived gene expression database for rice. J. Genet. Genom. 2017, 44, 235–241.
- Ferrari, C.; Proost, S.; Ruprecht, C.; Mutwil, M. PhytoNet: Comparative co-expression network analyses across phytoplankton and land plants. Nucleic Acids Res. 2018, 46, W76–W83.
- Proost, S.; Mutwil, M. CoNekT: An open-source framework for comparative genomic and transcriptomic network analyses. Nucleic Acids Res. 2018, 46, W133–W140.
- Lee, J.; Shah, M.; Ballouz, S.; Crow, M.; Gillis, J. CoCoCoNet: Conserved and comparative co-expression across a diverse set of species. Nucleic Acids Res. 2020, 48, W566–W571.
- Saithong, T.; Rongsirikul, O.; Kalapanulak, S.; Chiewchankaset, P.; Siriwat, W.; Netrphan, S.; Suksangpanomrung, M.; Meechai, A.; Cheevadhanarak, S. Starch biosynthesis in cassava: A genome-based pathway reconstruction and its exploitation in data integration. BMC Syst. Biol. 2013, 7, 75.
- Töpfer, N.; Caldana, C.; Grimbs, S.; Willmitzer, L.; Fernie, A.R.; Nikoloski, Z. Integration of Genome-Scale Modeling and Transcript Profiling Reveals Metabolic Pathways Underlying Light and Temperature Acclimation in Arabidopsis. Plant Cell 2013, 25, 1197–1211.
- Dharmawardhana, P.; Ren, L.; Amarasinghe, V.; Monaco, M.; Thomason, J.; Ravenscroft, D.; McCouch, S.; Ware, D.; Jaiswal, P. A genome scale metabolic network for rice and accompanying analysis of tryptophan, auxin and serotonin biosynthesis regulation under biotic stress. Rice 2013, 6, 15.
- Assefa, T.; Otyama, P.I.; Brown, A.V.; Kalberer, S.R.; Kulkarni, R.S.; Cannon, S.B. Genome-wide associations and epistatic interactions for internode number, plant height, seed weight and seed yield in soybean. BMC Genom. 2019, 20, 527.
- Weiße, A.Y.; Oyarzún, D.A.; Danos, V.; Swain, P.S. Mechanistic links between cellular trade-offs, gene expression, and growth. Proc. Natl. Acad. Sci. USA 2015, 112, E1038–E1047.
- Tang, S.; Zhao, H.; Lu, S.; Yu, L.; Zhang, G.; Zhang, Y.; Yang, Q.Y.; Zhou, Y.; Wang, X.; Ma, W.; et al. Genome- and transcrip-tome-wide association studies provide insights into the genetic basis of natural variation of seed oil content in Brassica napus. Mol. Plant 2021, 14, 470–487.
- Gusev, A.; Ko, A.; Shi, H.; Bhatia, G.; Chung, W.; Penninx, B.W.J.H.; Jansen, R.; de Geus, E.J.C.; Boomsma, D.I.; Wright, F.A.; et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 2016, 48, 245–252.
- Kremling, K.A.G.; Diepenbrock, C.H.; Gore, M.A.; Buckler, E.S.; Bandillo, N.B. Transcriptome-Wide Association Supplements Genome-Wide Association in Zea mays. G3 Genes Genomes Genet. 2019, 9, 3023–3033.
- Wu, D.; Li, X.; Tanaka, R.; Wood, J.C.; Tibbs-Cortes, L.E.; Magallanes-Lundback, M.; Bornowski, N.; Hamilton, J.P.; Vaillancourt, B.; Diepenbrock, C.H.; et al. Combining GWAS and TWAS to identify candidate causal genes for tocochromanol levels in maize grain. Genetics 2022, 221.
More
Information
Subjects:
Plant Sciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.5K
Revisions:
2 times
(View History)
Update Date:
25 Oct 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No