Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Patricia Dubot | -- | 2768 | 2022-10-24 17:57:52 | | | |
| 2 | Camila Xu | -4 word(s) | 2764 | 2022-10-25 04:11:19 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Dubot, P.; Astudillo, L.; Therville, N.; Carrié, L.; Pettazzoni, M.; Cheillan, D.; Stirnemann, J.; Levade, T.; Andrieu-Abadie, N.; Sabourdy, F. Cancer Prevalence in Sphingolipid Storage Disorders. Encyclopedia. Available online: https://encyclopedia.pub/entry/30981 (accessed on 23 July 2026).
Dubot P, Astudillo L, Therville N, Carrié L, Pettazzoni M, Cheillan D, et al. Cancer Prevalence in Sphingolipid Storage Disorders. Encyclopedia. Available at: https://encyclopedia.pub/entry/30981. Accessed July 23, 2026.
Dubot, Patricia, Leonardo Astudillo, Nicole Therville, Lorry Carrié, Magali Pettazzoni, David Cheillan, Jérôme Stirnemann, Thierry Levade, Nathalie Andrieu-Abadie, Frédérique Sabourdy. "Cancer Prevalence in Sphingolipid Storage Disorders" Encyclopedia, https://encyclopedia.pub/entry/30981 (accessed July 23, 2026).
Dubot, P., Astudillo, L., Therville, N., Carrié, L., Pettazzoni, M., Cheillan, D., Stirnemann, J., Levade, T., Andrieu-Abadie, N., & Sabourdy, F. (2022, October 24). Cancer Prevalence in Sphingolipid Storage Disorders. In Encyclopedia. https://encyclopedia.pub/entry/30981
Dubot, Patricia, et al. "Cancer Prevalence in Sphingolipid Storage Disorders." Encyclopedia. Web. 24 October, 2022.
Copy Citation
Sphingolipidoses are a subgroup of rare inherited disorders of lipid metabolism, most often due to a lysosomal enzymatic defect affecting sphingolipid catabolism. They are characterized by the accumulation of sphingolipids and their deacylated derivatives, called lysosphingolipids.
cancer
sphingolipid
glucosylsphingosine
Gaucher disease
1. Introduction
Sphingolipids (SLs) comprise a very large number of amphiphilic lipid molecules, having in common a long chain aminoalcohol referred to as a sphingoid base. They are present in plants and animals, particularly in mammals. Owing to their chemical structure, most of them are constituents of cell membranes and preferentially locate in the outer leaflet of the plasma membrane. Similar to diacylglycerolipids, some of the major SLs are also components of circulating lipoproteins. Of note, sphingosine 1-phosphate (S1P) is typically found in the extracellular compartment, being relatively abundant in plasma and lymph. Membrane SLs, which are, by far, the predominant SL species, contain one molecule of fatty acid. In accordance with previous classifications of lipids, where the term “lyso” denotes a glycero(phospho)lipid lacking a radyl group [1], lysosphingolipids (lysoSLs) are defined as SLs that lack the fatty acyl moiety.
SLs are synthesized by all mammalian cells. Their biosynthesis starts in the endoplasmic reticulum (ER) by the condensation of an acyl-CoA, mainly palmitoyl-CoA, and L-serine to form the backbone of the sphingoid base. Reduction of this initial product leads to sphinganine, which is N-acylated, i.e., linked to a fatty acyl moiety through an amide bond by a ceramide synthase. After desaturation, the resulting ceramide molecule can be further transformed into complex SLs, such as sphingomyelin or glycosphingolipids. Whereas sphingomyelin and β-glucosylceramide-based glycolipids are synthesized in the Golgi apparatus, β-galactosylceramide is formed in the ER. They all are transported to the plasma membrane. The degradation of diet-derived SLs is mediated by secreted intestinal enzymes. As to cellular SLs, their physiological catabolism mostly occurs in the lysosomes through a conserved sequence of hydrolytic steps (see Figure 1). In this pathway, the breakdown of complex SLs gradually releases the residues of the hydrophilic headgroup attached to the ceramide backbone. The last step of lysosomal degradation is catalyzed by acid ceramidase, which cleaves the amide bond and, thus, liberates sphingosine and fatty acids.
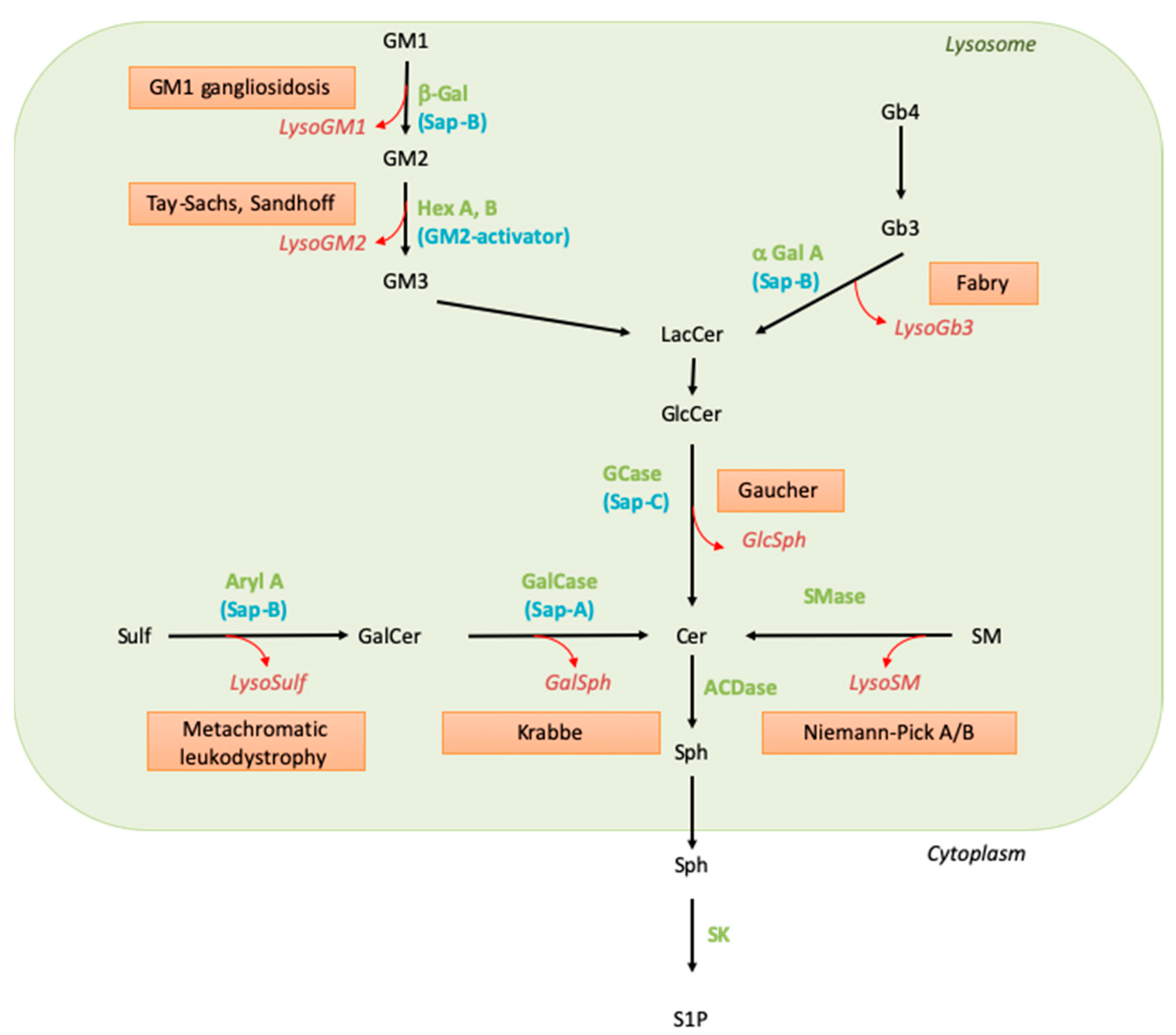
Figure 1. Sphingolipid catabolism and associated diseases. Green names correspond to the enzyme names, blue names to their activators, the red boxed texts contain the disease name, and the red and italic names indicate the corresponding lysoSL. Abbreviations: α-Gal: alpha-galactosidase; ACDase, acid ceramidase; Aryl A, arylsulfatase A; β-Gal, beta-galactosidase; Cer, ceramide; GalCase, galactosylceramidase; GalSph, galactosylsphingosine; GCase, glucosylceramidase; GlcCer, glucosylceramide; GlcSph, glucosylsphingosine; Hex, hexosaminidase; LacCer, lactosylceramide; LysoSulf, lysosulfatide; Sap, saposin; SK, sphingosine kinase; SM, sphingomyelin; SMase, sphingomyelinase; Sph, sphingosine; Sulf, sulfatide; S1P, sphingosine 1-phosphate.
Knowledge of SLs and SL metabolism has been strongly stimulated by the existence in humans of genetic conditions characterized by the accumulation of selective undegraded SL molecules. Extensive biochemical and genetic studies on these diseases, named sphingolipid storage disorders (or sphingolipidoses), led to the identification of their underlying defects more than 50 years ago. These disorders result from the disruption of the lysosomal catabolic pathway due to the deficient function of one of the hydrolases, either because this enzyme or its so-called activator protein (sphingolipid activator protein) is mutated, leading to profoundly decreased catalytic activity [2][3]. The catabolic pathway implicating these enzymatic and activator proteins is depicted in Figure 1, and the lipid molecules that accumulate in the corresponding sphingolipid storage disorders are listed in Table 1. As the entry of SLs into the endolysosomal compartment is permanent, although variable, depending on cell type and external conditions, the enzymatic defect translates into substrate accumulation in the lysosomes (see Table 1). Abnormally elevated concentrations of the undegraded SLs can be observed in organs as well as in biological fluids such as blood and urine; the presence of the accumulated SLs in the extracellular milieu may be explained by cell damage or lysosomal exocytosis.
Table 1. Primary accumulated SLs and lysoSLs in sphingolipid storage diseases. Of note, genetic conditions in which the accumulation of some SLs occurs as a secondary phenomenon are not listed here. The plasma concentrations of lysosphingolipids are indicated in brackets (control values).
| Sphingolipid Storage Disease | Affected Gene and Protein | Primary Stored Sphingolipid | Major Accumulated Lysosphingolipid | Lysosphingolipid Plasma Concentration (nmol/L) | Ref. |
|---|---|---|---|---|---|
| GM1-gangliosidosis | GLB1, GM1 β-galactosidase | GM1-ganglioside | LysoGM1 | 0–40 (<0.07) | [4][5] |
| GM2-gangliosidosis type B (Tay-Sachs disease) | HEXA, α subunit of β-hexosaminidase A | GM2-ganglioside | LysoGM2 | 0–14.5 (ND) | [5][6] |
| GM2-gangliosidosis type 0 (Sandhoff disease) | HEXB, β subunit of hexosaminidase A | GM2-ganglioside | LysoGM2 | 0–118 (ND) | [4][5] |
| GM2-gangliosidosis type AB | GM2A, GM2 activator protein | GM2-ganglioside | LysoGM2 * | NA | [7] |
| Gaucher disease | GBA, acid β-glucosidase (β-glucosylceramidase) |
Glucosylceramide | Glucosylsphingosine | 46–427 (<3.5) | [5][8][9] |
| Gaucher disease, saposin C deficiency | PSAP, prosaposin | Glucosylceramide | Glucosylsphingosine | 110 (<3) | [10] |
| Fabry disease | GLA, α-galactosidase A | Gb3 (trihexosylceramide) | LysoGb3 (globotriaosylsphingosine) |
0.5–150 (<0.6) | [5][11] |
| Metachromatic leukodystrophy | ARSA, arylsulfatase A | Sulfatide | Lysosulfatide | ND | [12][13][14] |
| Metachromatic leukodystrophy, saposin B deficiency | PSAP, prosaposin | Sulfatide | Lysosulfatide * | [15] | |
| Krabbe disease (globoid cell leukodystrophy) | GALC, β-galactosylceramidase | Galactosylceramide | Galactosylsphingosine (psychosine) | 1.5–54 (<2) | [16][17] |
| Krabbe disease, saposin A deficiency | PSAP, prosaposin | Galactosylceramide | Galactosylsphingosine (psychosine) | 12 (<3) # | [18] |
| Prosaposin deficiency | PSAP, proposin | Multiple SLs | Glucosylsphingosine, lysoGb3, lysoSM inconstantly | GlSph: 53–61 (<3) LysoGb3: 5–8 (<0.6) LysoSM: 15–22 (<15) GalSph: 0.9–1.7 (<1) |
[19] |
| Niemann–Pick disease (types A and B) | SMPD1, acid sphingomyelinase | Sphingomyelin | LysoSM (sphingosylphosphocholine) and PPCS (“lysoSM509”) | lysoSM: 8–70 (<2.6) LysoSM509: 127–364 (<9) |
[19][20] |
Abbreviations: LysoSM, lysosphingomyelin; PPCS, N-palmitoyl-O-phosphocholineserine. NA: not available. ND: not detectable. The asterisk (*) denotes that accumulation of the lysoSL is likely but, to researchers' knowledge, not yet demonstrated in patients. The symbol # indicates that the concentration was determined on dried blood spots.
The clinical presentation of patients affected with sphingolipid storage disorders is quite diverse, ranging from isolated visceral symptomatology to severe and fatal neuronopathic forms. A factor that determines organ involvement is the tissue distribution of the accumulated SLs. For instance, a neurological disease develops when the metabolism of GM1 and GM2 gangliosides or sulfatides and galactosylceramide, which are key components of neurons and myelin, respectively, are not properly degraded. On the other hand, the defective turnover of glucosylceramide (GlcCer) or sphingomyelin (SM), which are widely distributed, affects cells such as professional phagocytes, i.e., monocytes-macrophages. Of importance for the discussion below, the age of disease onset can vary widely, ranging from the antenatal period up to late adulthood. This variability is explained, at least partially, by the residual activity of the affected enzyme: the lower the activity is, the more severe the lipid storage and the symptomatology are [21].
As already mentioned, the deficient activity of a lysosomal hydrolase results in the accumulation of its primary substrate. However, biochemical studies performed as early as in the mid-seventies have also documented the presence and storage of deacylated SLs, e.g., galactosylsphingosine (GalSph), also called psychosine, and glucosylsphingosine (GlcSph), in organs of patients affected with Krabbe and Gaucher diseases, respectively [8][22]. The discovery of these two lysoSLs was soon followed by the observation that other similarly N-deacylated SLs accumulate in distinct sphingolipidoses (see Table 1). Abnormally high levels of lysoSLs are also found in patients’ biological fluids such as plasma, urine, and cerebrospinal fluid. The metabolic source of lysoSLs has long been debated. Initially, it was postulated that the synthesis of a lysoSL follows the same biosynthetic route as the conventional lipid but uses a sphingoid base rather than ceramide. Recent evidence has shown that instead, GalSph, GlcSph and lysoGb3 are generated by the action of acid ceramidase on their corresponding SL in the lysosomal compartment, i.e., where the parental lipid is stored [23][24]. In fact, the elimination of GalSph production by ablating acid ceramidase in a mouse model of Krabbe suppressed the behavioral and histopathological features of leukodystrophy [25]. Whether other lysoSLs are produced by acid ceramidase still requires further investigation. The fatty acid amide hydrolase (FAAH) enzyme has been reported to partially account for the production of lysosulfatide [26].
Finally, the present research, one should recall that sphingosine 1-phosphate (S1P) is also a lysoSL. This lysophospholipid is mainly formed by the sphingosine-kinase-mediated phosphorylation of sphingosine and by the autotaxin-mediated breakdown of lysoSM [27]. It is still unknown whether a ceramidase can generate S1P from ceramide 1-phosphate. So far, with the possible exception of Gaucher disease, neither accumulation of S1P nor ceramide 1-phosphate has been reported in sphingolipid storage disorders.
2. Cancer Prevalence in Sphingolipid Storage Disorders
Gaucher disease (GD) is the most frequent sphingolipid storage disease, of which the deficiency of lysosomal glucosylceramidase (GCase), encoded by the GBA gene, results in an accumulation of GlcCer and GlcSph (Table 1). Three disease forms are described: type 1 is the most frequent, with hepatosplenomegaly, anemia, thrombocytopenia, bone abnormalities, and lung complications without neurological involvement, the latter being only present in types 2 and 3 [28] (Table 2). Numerous case reports, case series, and cohort studies have described the occurrence of malignancies in Gaucher patients and have suggested that the risk of developing cancer is increased in Gaucher patients with a relative risk evaluated at 1.7 (95% confidence interval (95% CI) 1.3–2.3) [29]. In the French Gaucher Disease Registry (FGDR) (n = 657 patients), researchers report that 27 patients presented malignancies (4.1%) [30]. Compared to data from the French population (obtained from the International Agency for Research on Cancer (https://gco.iarc.fr/today/home) accessed on 31 March 2021), researchers observations suggest a marked increase in the prevalence of malignancies at an odds ratio (OR) of 1.8 (95% CI 1.03–2.81) (Table 3). The risk of multiple myeloma seems to be particularly marked, with an OR of 23.8 (95% CI 4.9–70.2) (see Table 2 and Table 3), which is very consistent with the literature [29][31][32][33]. Nevertheless, these results should be interpreted with caution because of limits relative to study design. In addition, an association with monoclonal gammopathy of undetermined significance (MGUS), a pre-malignant condition for multiple myeloma, is also suspected [32]. In the FGDR, MGUS has been found in 59 patients (25%) [34]. Besides the risk of hematological malignancies, the prevalence of digestive cancer and, especially, hepatocellular carcinoma in Gaucher patients also seems to be increased [35]. However, in the FGDR, no hepatocellular carcinoma has been observed. Interestingly, in the FGDR, five Gaucher patients presented two cancers, a finding that has been previously described [31][33][36] (see Table 3).
Table 2. Overview of malignancies described in patients affected with Gaucher disease, Fabry disease, Niemann–Pick B disease, or metachromatic leukodystrophy.
| Sphingolipid Storage Disease | Clinical Presentation/Affected Organs | Benign and Precancerous Lesions | Malignant Tumors | |
|---|---|---|---|---|
| Organ system | Sign/symptom(s) | |||
| Gaucher disease OMIM#230800 [37] |
Blood | Anemia, thrombopenia | Dysgammaglobulinemia-MGUS [29][34][38] | Hematological malignancies: [29][31][32][33]
|
| Viscera | Hepatosplenomegaly, gallstones | Digestive cancers
|
||
| Bone | Bone pain, bone infarcts, avascular necrosis, pseudo-osteomyelitis | |||
| Lung | Interstitial disease, fibrosis | Lung cancer [31][41][42] | ||
| Skin | Collodion baby | Skin cancer:
|
||
| Thyroid cancer [31][36][40] | ||||
| Fabry disease OMIM #301500 [43] |
Nervous system | Periodic crises of acroparesthesia, sweating abnormalities |
Meningioma [44][45] | |
| Cerebrovascular disease | Stroke, transient ischemic attack | |||
| Eyes | Cornea verticillata | |||
| Skin | Angiokeratoma | Melanoma [45][46] | ||
| Kidney | Nephropathy to end-stage renal disease | Renal cell carcinoma [45][46][47][48][49] | ||
| Heart | Cardiac damage: left ventricular hypertrophy, cardiomyopathy, arrhythmia | |||
| Gastrointestinal tract | Nausea, vomiting, diarrhea | Colon polyp [45] | Colon cancer [50] | |
| Blood | MGUS [45] | Blood cancers [51][52] | ||
| Niemann–Pick B OMIM #607616 |
Viscera | Progressive hepatosplenomegaly, deterioration in liver function | Liver cancer [53] | |
| Blood | Thrombocytopenia | MGUS [54] | Multiple myeloma [53][55] | |
| Lung | Interstitial disease | Lung cancer [54] | ||
| Bone | Osteopenia | Chondrosarcoma [53] | ||
| Bladder cancer [54] Breast cancer [54] Thyroid cancer [54] |
||||
| Metachromatic leukodystrophy OMIM #250100 |
Brain | Progressive neurological damage with leukodystrophy | ||
| Gallbladder | Hemobilia | Gallbladder polyposis [56] | Gallbladder carcinoma [57][58][59] | |
Table 3. Cancer prevalence in the French Gaucher Disease Registry and French population.
| French Gaucher Disease Registry (n = 445 Living Patients) |
French Population, All Ages, Both Genders (International Agency for Research on Cancer, www.gco.iarc.fr, Accessed on 31 March 2021) (n = 65,273,512 Living People) |
Odds Ratio, 95%CI, p-Value (Fisher’s Exact Test) |
|
|---|---|---|---|
| Cancer Type | Living Patients Number | Living Patients Number | |
| All Cancers | 18 | 1,501,881 | 1.8 [1.03–2.81], p < 0.05 |
| Blood cancers (all) | 6 | 104,838 | 8.4 [3.1–18.4], p < 0.0001 |
|
3 | 18,442 | 23.8 [4.9–70.2], p < 0.0001 |
|
2 | 44,809 | 6.5 [0.8–23.8], p < 0.05 |
|
1 | NA | NC |
Digestive Cancer
|
2 | 86,328 | 3.4 [0.4–12.3], p = 0.11 |
| Lung Cancer | 2 | 59,708 | 4.9 [0.6–17.9], p = 0.06 |
Skin Cancer
|
3 3 |
316,830 260,694 |
1.4 [0.3–4.1], p < 0.5 |
| Thyroid Cancer | 2 | 50,301 | 5.8 [0.7–21.2], p < 0.05 |
Gynaecological Cancer
|
3 | 251,161 | 1.8 [0.4–5.2], p = 0.25 |
Urological cancer
|
2 | 312,121 | 0.9 [0.1–3.4], p = 1 |
NA: not available; NC: not calculated.
All these observations would suggest that GD could be a predisposing condition for the development of cancers.
The risk of cancer could also be increased in patients suffering from other sphingolipidoses. Indeed, some case reports [44][47][48][49][50][51][52] and cohort studies [45][46] have also described the development of cancer in Fabry patients. Fabry disease (FD), the second most frequent sphingolipid storage disease, is an X-linked disorder resulting from the deficient activity of lysosomal alpha-galactosidase A, encoded by the GLA gene, leading to an accumulation of Gb3 and lysoGb3 (Table 1). FD is classically characterized by angiokeratoma, cornea verticillata, neuropathy (acroparesthesia), progression toward chronic kidney disease, cerebrovascular accidents, and hypertrophic cardiomyopathy (Table 2). In British (n = 261) [45] and Italian (n = 53) [60] cohorts of Fabry patients, an increased incidence was observed for urological and kidney cancers, as well as melanoma, compared to the general population [45]. An increase in the occurrence of meningioma, a benign tumor, has also been observed (see Table 2) [45][46].
The occurrence of cancer in Niemann–Pick disease types A and B (NPD-A and B) could also be non-negligible. NPD-A and -B are very rare diseases that result from the deficient activity of acid sphingomyelinase (ASM), encoded by the SMPD1 gene, leading to an accumulation of SM and lysoSM (Table 1). Clinical phenotypes range from a very severe phenotype (NPD-A), with neurovisceral involvement and an early death, to a mild phenotype (NPD-B), with hepatosplenomegaly, cytopenia, and lung involvement. Very recently, an increase in the prevalence of cancer has been shown in a French cohort of adults (n = 31) suffering from NPD-A and -B, in which five patients presented with cancer, two of whom had lung cancer, and two patients presented MGUS (Table 2) [54]. In addition, cancer has been reported as the cause of death in 9% of patients with NPD-B [53].
To a lesser extent, metachromatic leukodystrophy (MLD) could be associated with gallbladder carcinoma. MLD is caused by the deficient activity of arylsulfatase A, encoded by the ARSA gene, which results in the accumulation of sulfatide and lysosulfatide (Table 1). Three clinical subtypes are described: the late-infantile (0–2.5 years), juvenile (2.5–16 years), and adult (>16 years) forms [61]. Patients with this disease present a progressive neurological involvement with leukodystrophy at MRI; gallbladder complications such as hemobilia and cholecystitis are also described [56] (Table 2). Interestingly, in a cohort of 34 patients, an increase in the prevalence of gallbladder polyposis, a precancerous condition for gallbladder carcinoma, has been observed [57]. In addition, cases of gallbladder carcinoma with liver metastases have been reported in two patients with an adult form of MLD [57][58] and a case without metastases in one patient with a juvenile form [59]. It should be noted that in these patients, the carcinoma occurred earlier than in the general population (56–63 years).
Taken together, these observations suggest a potential link between the occurrence of cancer and the above four sphingolipid storage diseases. In some cases, a common location was observed between typically affected organs and associated cancer, e.g., cytopenia and blood cancers in GD or chronic kidney disease and kidney cancer in FD (Table 2). It seems that there is no correlation between the plasma concentration of lysoSLs and the risk of developing malignancies. The development of cancer seems to be associated with mild or late-onset forms of sphingolipid storage diseases. For instance, the majority of Gaucher patients having cancer present the classical mild form type 1. Indeed, a very slight augmentation of plasma GlcSph levels is associated with the occurrence of GlcSph-reactive IgGs [62]. Moreover, in the studied cohorts of Fabry patients with cancer, there are fewer males than females [45][46]. Females usually have lower lysoGb3 levels, present a milder, late-onset form of the disease, and very rarely develop kidney complications compared to males. No patient who is affected by a severe form of NPD and suffers from cancer has been reported [53][54]. These observations could be explained by life expectancy, which is longer in the milder forms, thus allowing cancer development. Moreover, the above findings were based on retrospective or cross-sectional studies, which limits the scientific evidence level. This would require further randomized studies in order to confirm the association between sphingolipid storage diseases and cancer and, if necessary, to determine risk factors such as splenectomy, treatment, SLs, and lysoSL levels.
Although the underlying mechanisms remain unknown, the accumulation of sphingolipids and their corresponding lysoSLs could be implicated in the increased risk of cancer. In GD, the therapeutic splenectomy may lead to the release of undegraded lipids and could be associated with the occurrence of multiple cancers in the same patient [36]. In the FGDR, three out of five patients with two distinct cancers were splenectomized (data were unavailable for the remaining two). A common treatment for Gaucher and Fabry patients is enzyme replacement therapy (ERT), which is based on the administration of exogenous recombinant enzymes. Considering that these treatments improve clinical symptoms and reduce circulating lysoSL levels, they would be expected to be protective against cancer development, which has already been reported [63]. However, reports in the literature are inconsistent in this regard [64], which may suggest that the build-up of these lipids could result in early irreversible protumor events.
References
- Fahy, E.; Subramaniam, S.; Brown, H.A.; Glass, C.K.; Merrill, A.H., Jr.; Murphy, R.C.; Raetz, C.R.; Russell, D.W.; Seyama, Y.; Shaw, W.; et al. A comprehensive classification system for lipids. J. Lipid Res. 2005, 46, 839–861.
- Kolter, T.; Sandhoff, K. Sphingolipid metabolism diseases. Biochim. Biophys. Acta 2006, 1758, 2057–2079.
- Breiden, B.; Sandhoff, K. Lysosomal Glycosphingolipid Storage Diseases. Annu. Rev. Biochem. 2019, 88, 461–485.
- Kobayashi, T.; Goto, I.; Okada, S.; Orii, T.; Ohno, K.; Nakano, T. Accumulation of lysosphingolipids in tissues from patients with GM1 and GM2 gangliosidoses. J. Neurochem. 1992, 59, 1452–1458.
- Pettazzoni, M.; Froissart, R.; Pagan, C.; Vanier, M.T.; Ruet, S.; Latour, P.; Guffon, N.; Fouilhoux, A.; Germain, D.P.; Levade, T.; et al. LC-MS/MS multiplex analysis of lysosphingolipids in plasma and amniotic fluid: A novel tool for the screening of sphingolipidoses and Niemann-Pick type C disease. PLoS ONE 2017, 12, e0181700.
- Neuenhofer, S.; Conzelmann, E.; Schwarzmann, G.; Egge, H.; Sandhoff, K. Occurrence of lysoganglioside lyso-GM2 (II3-Neu5Ac-gangliotriaosylsphingosine) in GM2 gangliosidosis brain. Biol. Chem. Hoppe Seyler 1986, 367, 241–244.
- Kodama, T.; Togawa, T.; Tsukimura, T.; Kawashima, I.; Matsuoka, K.; Kitakaze, K.; Tsuji, D.; Itoh, K.; Ishida, Y.; Suzuki, M.; et al. Lyso-GM2 ganglioside: A possible biomarker of Tay-Sachs disease and Sandhoff disease. PLoS ONE 2011, 6, e29074.
- Raghavan, S.S.; Mumford, R.A.; Kanfer, J.N. Isolation and characterization of glucosylsphingosine from Gaucher’s spleen. J. Lipid Res. 1974, 15, 484–490.
- Nilsson, O.; Mansson, J.-E.; Hakansson, G.; Svennerholm, L. The occurrence of psychosine and other glycolipids in spleen and liver from the three major types of Gaucher’s disease. Biochim. Biophys. Acta Lipids Lipid Metab. 1982, 712, 453–463.
- Kang, L.; Zhan, X.; Ye, J.; Han, L.; Qiu, W.; Gu, X.; Zhang, H. A rare form of Gaucher disease resulting from saposin C deficiency. Blood Cells Mol. Dis. 2018, 68, 60–65.
- Aerts, J.M.; Groener, J.E.; Kuiper, S.; Donker-Koopman, W.E.; Strijland, A.; Ottenhoff, R.; van Roomen, C.; Mirzaian, M.; Wijburg, F.A.; Linthorst, G.E.; et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Natl. Acad. Sci. USA 2008, 105, 2812–2817.
- Toda, K.; Kobayashi, T.; Goto, I.; Kurokawa, T.; Ogomori, K. Accumulation of lysosulfatide (sulfogalactosylsphingosine) in tissues of a boy with metachromatic leukodystrophy. Biochem. Biophys. Res. Commun. 1989, 159, 605–611.
- Rosengren, B.; Fredman, P.; Månsson, J.E.; Svennerholm, L. Lysosulfatide (galactosylsphingosine-3-O-sulfate) from metachromatic leukodystrophy and normal human brain. J. Neurochem. 1989, 52, 1035–1041.
- Saville, J.T.; Smith, N.J.; Fletcher, J.M.; Fuller, M. Quantification of plasma sulfatides by mass spectrometry: Utility for metachromatic leukodystrophy. Anal. Chim. Acta 2017, 955, 79–85.
- Deconinck, N.; Messaaoui, A.; Ziereisen, F.; Kadhim, H.; Sznajer, Y.; Pelc, K.; Nassogne, M.C.; Vanier, M.T.; Dan, B. Metachromatic leukodystrophy without arylsulfatase A deficiency: A new case of saposin-B deficiency. Eur. J. Paediatr. Neurol. 2008, 12, 46–50.
- Jain, M.; De Jesus, O. Krabbe Disease. In StatPearls; StatPearls Publishing Copyright © 2022, StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022.
- Zhao, S.; Zhan, X.; Wang, Y.; Ye, J.; Han, L.; Qiu, W.; Gao, X.; Gu, X.; Zhang, H. Large-scale study of clinical and biochemical characteristics of Chinese patients diagnosed with Krabbe disease. Clin. Genet. 2018, 93, 248–254.
- Calderwood, L.; Wenger, D.A.; Matern, D.; Dahmoush, H.; Watiker, V.; Lee, C. Rare Saposin A deficiency: Novel variant and psychosine analysis. Mol. Genet. Metab. 2020, 129, 161–164.
- Motta, M.; Tatti, M.; Furlan, F.; Celato, A.; Di Fruscio, G.; Polo, G.; Manara, R.; Nigro, V.; Tartaglia, M.; Burlina, A.; et al. Clinical, biochemical and molecular characterization of prosaposin deficiency. Clin. Genet. 2016, 90, 220–229.
- Kuchar, L.; Sikora, J.; Gulinello, M.E.; Poupetova, H.; Lugowska, A.; Malinova, V.; Jahnova, H.; Asfaw, B.; Ledvinova, J. Quantitation of plasmatic lysosphingomyelin and lysosphingomyelin-509 for differential screening of Niemann-Pick A/B and C diseases. Anal. Biochem. 2017, 525, 73–77.
- Leinekugel, P.; Michel, S.; Conzelmann, E.; Sandhoff, K. Quantitative correlation between the residual activity of beta-hexosaminidase A and arylsulfatase A and the severity of the resulting lysosomal storage disease. Hum. Genet. 1992, 88, 513–523.
- Vanier, M.; Svennerholm, L. Chemical pathology of Krabbe disease: The occurrence of psychosine and other neutral sphingoglycolipids. Adv. Exp. Med. Biol. 1976, 68, 115–126.
- Yamaguchi, Y.; Sasagasako, N.; Goto, I.; Kobayashi, T. The synthetic pathway for glucosylsphingosine in cultured fibroblasts. J. Biochem. 1994, 116, 704–710.
- Ferraz, M.J.; Marques, A.R.; Appelman, M.D.; Verhoek, M.; Strijland, A.; Mirzaian, M.; Scheij, S.; Ouairy, C.M.; Lahav, D.; Wisse, P.; et al. Lysosomal glycosphingolipid catabolism by acid ceramidase: Formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett. 2016, 590, 716–725.
- Li, Y.; Xu, Y.; Benitez, B.A.; Nagree, M.S.; Dearborn, J.T.; Jiang, X.; Guzman, M.A.; Woloszynek, J.C.; Giaramita, A.; Yip, B.K.; et al. Genetic ablation of acid ceramidase in Krabbe disease confirms the psychosine hypothesis and identifies a new therapeutic target. Proc. Natl. Acad. Sci. USA 2019, 116, 20097–20103.
- Yaghootfam, C.; Gehrig, B.; Sylvester, M.; Gieselmann, V.; Matzner, U. Deletion of fatty acid amide hydrolase reduces lyso-sulfatide levels but exacerbates metachromatic leukodystrophy in mice. J. Biol. Chem. 2021, 297, 101064.
- Clair, T.; Aoki, J.; Koh, E.; Bandle, R.W.; Nam, S.W.; Ptaszynska, M.M.; Mills, G.B.; Schiffmann, E.; Liotta, L.A.; Stracke, M.L. Autotaxin hydrolyzes sphingosylphosphorylcholine to produce the regulator of migration, sphingosine-1-phosphate. Cancer Res. 2003, 63, 5446–5453.
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441.
- Arends, M.; van Dussen, L.; Biegstraaten, M.; Hollak, C.E.M. Malignancies and monoclonal gammopathy in Gaucher disease; a systematic review of the literature. Br. J. Haematol. 2013, 161, 832–842.
- Dubot, P.; Astudillo, L.; Therville, N.; Sabourdy, F.; Stirnemann, J.; Levade, T.; Andrieu-Abadie, N. Are Glucosylceramide-Related Sphingolipids Involved in the Increased Risk for Cancer in Gaucher Disease Patients? Review and Hypotheses. Cancers 2020, 12, 475.
- Taddei, T.H.; Kacena, K.A.; Yang, M.; Yang, R.; Malhotra, A.; Boxer, M.; Aleck, K.A.; Rennert, G.; Pastores, G.M.; Mistry, P.K. The underrecognized progressive nature of N370S Gaucher disease and assessment of cancer risk in 403 patients. Am. J. Hematol. 2009, 84, 208–214.
- de Fost, M.; Vom Dahl, S.; Weverling, G.J.; Brill, N.; Brett, S.; Häussinger, D.; Hollak, C.E.M. Increased incidence of cancer in adult Gaucher disease in Western Europe. Blood Cells Mol. Dis. 2006, 36, 53–58.
- Landgren, O.; Turesson, I.; Gridley, G.; Caporaso, N.E. Risk of malignant disease among 1525 adult male US Veterans with Gaucher disease. Arch. Intern. Med. 2007, 167, 1189–1194.
- Nguyen, Y.; Stirnemann, J.; Lautredoux, F.; Cador, B.; Bengherbia, M.; Yousfi, K.; Hamroun, D.; Astudillo, L.; Billette de Villemeur, T.; Brassier, A.; et al. Immunoglobulin Abnormalities in Gaucher Disease: An Analysis of 278 Patients Included in the French Gaucher Disease Registry. Int. J. Mol. Sci. 2020, 21, 1247.
- Regenboog, M.; van Dussen, L.; Verheij, J.; Weinreb, N.J.; Santosa, D.; Vom Dahl, S.; Häussinger, D.; Müller, M.N.; Canbay, A.; Rigoldi, M.; et al. Hepatocellular carcinoma in Gaucher disease: An international case series. J. Inherit. Metab. Dis. 2018, 41, 819–827.
- Lo, S.M.; Stein, P.; Mullaly, S.; Bar, M.; Jain, D.; Pastores, G.M.; Mistry, P.K. Expanding spectrum of the association between Type 1 Gaucher disease and cancers: A series of patients with up to 3 sequential cancers of multiple types--correlation with genotype and phenotype. Am. J. Hematol. 2010, 85, 340–345.
- Stirnemann, J.; Vigan, M.; Hamroun, D.; Heraoui, D.; Rossi-Semerano, L.; Berger, M.G.; Rose, C.; Camou, F.; de Roux-Serratrice, C.; Grosbois, B.; et al. The French Gaucher’s disease registry: Clinical characteristics, complications and treatment of 562 patients. Orphanet J. Rare Dis. 2012, 7, 77.
- Jurecka, A.; Gregorek, H.; Kleinotiene, G.; Czartoryska, B.; Tylki-Szymanska, A. Gaucher disease and dysgammaglobulinemia: a report of 61 patients, including 18 with GD type III. Blood Cells Mol. Dis. 2011, 46, 85–87.
- Lee, R.E. The pathology of Gaucher disease. Prog. Clin. Biol. Res. 1982, 95, 177–217.
- Zimran, A.; Liphshitz, I.; Barchana, M.; Abrahamov, A.; Elstein, D. Incidence of malignancies among patients with type I Gaucher disease from a single referral clinic. Blood Cells Mol. Dis. 2005, 34, 197–200.
- Leone, J.P.; Dudek, A.Z. Enzyme replacement therapy for Gaucher’s disease in patient treated for non-small cell lung cancer. Anticancer Res. 2008, 28, 3937–3939.
- Shuai, W.; Wagner, C.E.; Sukswai, N.; Medeiros, L.J.; Bueso-Ramos, C.; Oo, T.H. Gaucher disease type 1 first recognized in an elderly patient with thrombocytopenia and lung adenocarcinoma. Clin. Case Rep. 2019, 7, 1804–1805.
- Germain, D.P. Fabry disease. Orphanet J Rare Dis 2010, 5, 30.
- Thurberg, B.L.; Germain, D.P.; Perretta, F.; Jurca-Simina, I.E.; Politei, J.M. Fabry disease: Four case reports of meningioma and a review of the literature on other malignancies. Mol. Genet. Metab. Rep. 2017, 11, 75–80.
- Bird, S.; Hadjimichael, E.; Mehta, A.; Ramaswami, U.; Hughes, D. Fabry disease and incidence of cancer. Orphanet J. Rare Dis. 2017, 12, 150.
- Rossi, F.; Auricchio, S.; Binaggia, A.; L’imperio, V.; Pagni, F.; Pieruzzi, F. Tumour incidence in Fabry disease: A cross-sectional study. J. Onco-nephrol. 2019, 3, 80–87.
- Pagni, F.; Pieruzzi, F.; Zannella, S.; Di Giacomo, A.; Bovo, G.; Ferrario, F.; Torti, G.; Rivera, R.; Assi, E.; Viglione, F.; et al. Possible pathogenetic relationship between Fabry disease and renal cell carcinoma. Am. J. Nephrol. 2012, 36, 537–541.
- Blanco, J.; Herrero, J.; Arias, L.F.; Garcia-Miralles, N.; Gamez, C.; Barrientos, A. Renal variant of Anderson-Fabry disease and bilateral renal cell carcinoma. Pathol. Res. Pract. 2005, 200, 857–860.
- Cassiman, D.; Claes, K.; Lerut, E.; Oyen, R.; Joniau, S.; Van Damme, B.; Jaeken, J. Bilateral renal cell carcinoma development in long-term Fabry disease. J. Inherit. Metab. Dis. 2007, 30, 830–831.
- Kusama, M.; Kimura, K.; Koyanagi, Y.; Tsuchida, A.; Yoshimatsu, A.; Ebinara, Y. A case report of atypical Fabry’s disease with colon cancer. Nihon Geka Gakkai Zasshi 1993, 94, 755–757.
- Tisi, M.C.; Zampetti, A.; Feliciani, C.; Fianchi, L.; Valentini, C.G.; Hohaus, S.; Larocca, L.M.; Leone, G.; Voso, M.T. Small lymphocytic lymphoma in a patient with Fabry disease. Leuk Lymphoma 2013, 54, 184–185.
- Cybulla, M.; Kleber, M.; Walter, K.N.; Kroeber, S.M.; Neumann, H.P.; Engelhardt, M. Is Fabry disease associated with leukaemia? Br. J. Haematol. 2006, 135, 264–265.
- Cassiman, D.; Packman, S.; Bembi, B.; Turkia, H.B.; Al-Sayed, M.; Schiff, M.; Imrie, J.; Mabe, P.; Takahashi, T.; Mengel, K.E.; et al. Cause of death in patients with chronic visceral and chronic neurovisceral acid sphingomyelinase deficiency (Niemann-Pick disease type B and B variant): Literature review and report of new cases. Mol. Genet. Metab. 2016, 118, 206–213.
- Mauhin, W.; Levade, T.; Vanier, M.T.; Froissart, R.; Lidove, O. Prevalence of Cancer in Acid Sphingomyelinase Deficiency. J. Clin. Med. 2021, 10, 5029.
- Portier, E.; Talbot, A.; Nguyen, Y.; Royer, B.; Pettazzoni, M.; Ben Salah, I.; Trichet, C.; Vercellino, L.; Arnulf, B.; Belmatoug, N. Multiple myeloma occurring in a case of Niemann-Pick disease Type B: A pathophysiological link? Br. J. Haematol. 2022, 197, e53–e55.
- Agarwal, A.; Shipman, P.J. Gallbladder polyposis in metachromatic leukodystrophy. Pediatr. Radiol. 2013, 43, 631–633.
- van Rappard, D.F.; Bugiani, M.; Boelens, J.J.; van der Steeg, A.F.; Daams, F.; de Meij, T.G.; van Doorn, M.M.; van Hasselt, P.M.; Gouma, D.J.; Verbeke, J.I.; et al. Gallbladder and the risk of polyps and carcinoma in metachromatic leukodystrophy. Neurology 2016, 87, 103–111.
- Simanovsky, N.; Ackerman, Z.; Kiderman, A.; Fields, S. Unusual gallbladder findings in two brothers with metachromatic leukodystrophy. Pediatr. Radiol. 1998, 28, 706–708.
- Koshu, K.; Ikeda, T.; Tamura, D.; Muramatsu, K.; Osaka, H.; Ono, S.; Adachi, K.; Nanba, E.; Nakajima, T.; Yamagata, T. Gallbladder cancer with ascites in a child with metachromatic leukodystrophy. Brain Dev. 2021, 43, 140–143.
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421.
- van Rappard, D.F.; Boelens, J.J.; Wolf, N.I. Metachromatic leukodystrophy: Disease spectrum and approaches for treatment. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 261–273.
- Allain-Maillet, S.; Bosseboeuf, A.; Mennesson, N.; Bostoën, M.; Dufeu, L.; Choi, E.H.; Cleyrat, C.; Mansier, O.; Lippert, E.; Le Bris, Y.; et al. Anti-Glucosylsphingosine Autoimmunity, JAK2V617F-Dependent Interleukin-1β and JAK2V617F-Independent Cytokines in Myeloproliferative Neoplasms. Cancers 2020, 12, 2446.
- Harel, R.; Gavish, I.; Aviv, A.; Greenman Maravi, N.; Trougouboff, P.; Zimran, A.; Revel-Vilk, S. Enzyme replacement therapy leading to improvement in myeloma indices in a patient with concomitant Gaucher disease. Intern. Med. J. 2022, 52, 872–875.
- Jaffe, D.H.; Flaks-Manov, N.; Benis, A.; Gabay, H.; DiBonaventura, M.; Rosenbaum, H.; Joseph, A.; Bachrach, A.; Leventer-Roberts, M. Population-based cohort of 500 patients with Gaucher disease in Israel. BMJ Open 2019, 9, e024251.
More
Information
Subjects:
Medicine, Research & Experimental
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
895
Revisions:
2 times
(View History)
Update Date:
25 Oct 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No