 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maria Grazia Giansanti | + 1179 word(s) | 1179 | 2020-02-03 07:36:21 | | | |
| 2 | Nicole Yin | + 9 word(s) | 1188 | 2020-10-27 03:46:00 | | |
Video Upload Options
Golgi phosphoprotein 3 (GOLPH3), a Phosphatidylinositol 4-Phosphate [PI(4)P] effector at the Golgi, is required for several intracellular functions, including Golgi ribbon structure maintenance, Golgi glycosylation and vesicle trafficking. It is amplified in several solid tumor types and its overexpression correlates with poor prognosis. GOLPH3 influences tumorigenesis through (i) regulation of Golgi-to-plasma membrane trafficking; (ii) turnover and glycosylation of cancer-relevant glycoproteins; (iii) influence on DNA damage response and maintenance of genomic stability.
1. Introduction
An increasingly number of reports in the last years demonstrated that intracellular vesicle trafficking deregulation is a feature of several cancers [1][2]. Golgi phosphoprotein 3 (GOLPH3) is the first recognized example of a Golgi resident oncoprotein[3] and it was originally identified in studies investigating the Golgi proteome[4][5]. GOLPH3 protein is highly conserved from yeast to humans, and localizes to the trans-Golgi via the direct interaction with PI(4)P; this localization is mediated by its unique C-terminal GPP34 domain[6][7] and is necessary to maintain Golgi architecture in human cells and Drosophila melanogaster[6][8].
2. Roles of GOLPH3, a PI(4)P Binding Protein, in the Physiopathology of Cancer
By using unbiased genome-wide copy number analysis, Scott and coauthors[9] identified a recurrent 5p13 amplification in several human solid tumors such as melanoma, colon adenocarcinoma and non-small-cell lung cancer. Four genes map within this chromosomal region and GOLPH3 was demonstrated to be the gene targeted for activation in cancers; in fact, its depletion is sufficient to revert oncogenic transformation in cell cultures. In addition, GOLPH3 cooperates with activated HRASV12 to increase transformed focus formation in Ink4a/Arf-deficient primary mouse embryonic fibroblasts[9]. Moreover, GOLPH3 up-regulation significantly enhances mouse xenograft tumor growth in vivo. Overall these data support the role of GOLPH3 as an oncogene[9]. Since this study, more than 30 research papers showed the overexpression of GOLPH3 in several cancers including melanoma, lung cancer, breast cancer, glioma, and colorectal cancer[10][11]. Several of them also provided evidence that GOLPH3 promotes cell proliferation and tumorigenicity of MDA-MB231 and MCF-7 breast cell lines and U251 and U87 glioblastoma cell lines[12][13][14][15]. Furthermore, it has been shown that overexpression of GOLPH3 is frequently associated with poor prognosis in multiple tumor types including 52% of breast cancers[13] and 41-53% of glioblastoma[14][15]. These findings support the role of GOLPH3 in cancer progression.
The oncogenic activity of GOLPH3 has been primarily correlated with its ability to influence signaling downstream of the mammalian target of rapamycin (mTOR), resulting in enhanced activation of both mammalian TORC1 and TORC2 complexes[9][10]. This idea has been recently supported by preclinical treatment studies showing that GOLPH3-expressing tumors are much more sensitive to mTOR inhibitors, suggesting that GOLPH3 levels might predict rapamycin efficacy in tumor therapy. However, the mechanistic basis that links the mTOR pathway to GOLPH3 remains to be elucidated, and multiple molecular mechanisms have been suggested to explain how GOLPH3 drives cancer[16][17].
The review of Sechi and co-workers[18] highlights the cellular processes that require GOLPH3 protein and the molecular pathways involved in GOLPH3-driven oncogenic transformation and metastasis in different types of tumors. It also reports the effects of using some chemotherapeutic drugs (such as gefitinib, 5-Fluorouracil or temozolomide), so that GOLPH3 knockdown might be exploited in combination with these substances to enhance the efficacy of chemotherapy in some tumor types.
Figure 1 shows the molecular mechanisms underlying GOLPH3 function in human cancer.
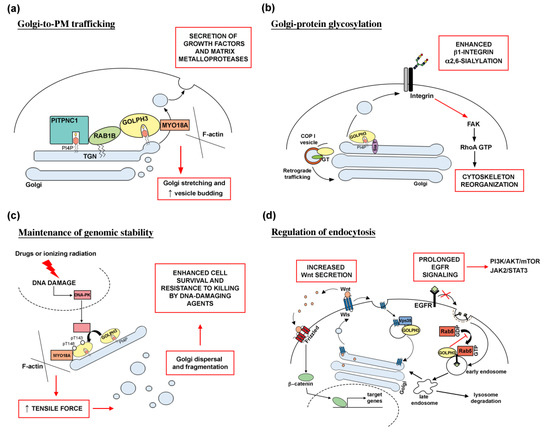
Figure 1. Molecular mechanisms underlying GOLPH3 function in human cancer. (a) Altered Golgi-to-PM trafficking. Recruitment of the PI(4)P-binding protein GOLPH3 at the trans-Golgi network (TGN), requires the PITPNC1/RAB1B network. Overactivation of the GOLPH3/MYO18A pathway leads to Golgi extension and enhanced vesicle release, resulting in the massive secretion of growth factors and matrix metalloproteases, which are responsible for cancer malignancy. (b) GOLPH3 role in Golgi-protein glycosylation. GOLPH3 is involved in COPI-mediated retrograde trafficking of several Golgi glycosyltransferases (GT). GOLPH3 protein binds to sialyltransferases (Sia T); its overexpression results in enhanced α2,6-sialylation of β1-integrins. Altered glycosylation affects integrin-mediated signaling pathway, leading to activation of FAK (focal adhesion kinase) and RhoA-dependent actin cytoskeleton reorganization and cell migration. (c) GOLPH3 is required for Golgi ribbon reorganization in response to DNA damage. DNA damage, induced by treatment with drugs and ionizing radiation, leads to activation of the DNA damage protein kinase (DNA-PK) that phosphorylates GOLPH3 on T143 and T148 residues. In turn, phosphorylation of GOLPH3 increases its interaction with MYO18A and its tensile force on the Golgi, leading to fragmentation of Golgi membranes and their dispersal throughout the cytoplasm. Massive Golgi fragmentation has been correlated to tumor development and maintenance since it is linked to cell survival and resistance to killing by DNA-damaging agents. (d) Relationship between GOLPH3 oncogenic activity and endocytic trafficking. GOLPH3 associates with the retromer complex subunit Vps35 to regulate recycling of several transmembrane receptors including the Wnt chaperone Wntless (Wls). In glioma cell lines, GOLPH3 overexpression increases Wls recycling and Wnt secretion, leading to abnormal Wnt/β-catenin signaling. GOLPH3 oncogenic activity in endocytic trafficking is responsible for a prolonged EGFR signaling. GOLPH3 binds to Rab5 and inhibits its activation on early endosome. High levels of GOLPH3 protein impair Rab5-mediated endocytosis and degradation of EGFR. As a result, overactivation of downstream signaling pathways such as PI3K/AKT/mTOR and JAK2/STAT3 promote cell proliferation and tumor transformation.
Figure 2 shows the effects of GOLPH3 up-regulation on solid tumors and the molecular pathways involved. As expected, the up-regulation of GOLPH3 is correlated with the up-regulation of other oncogenes or with the down-regulation of oncosuppressors (or related pathways).
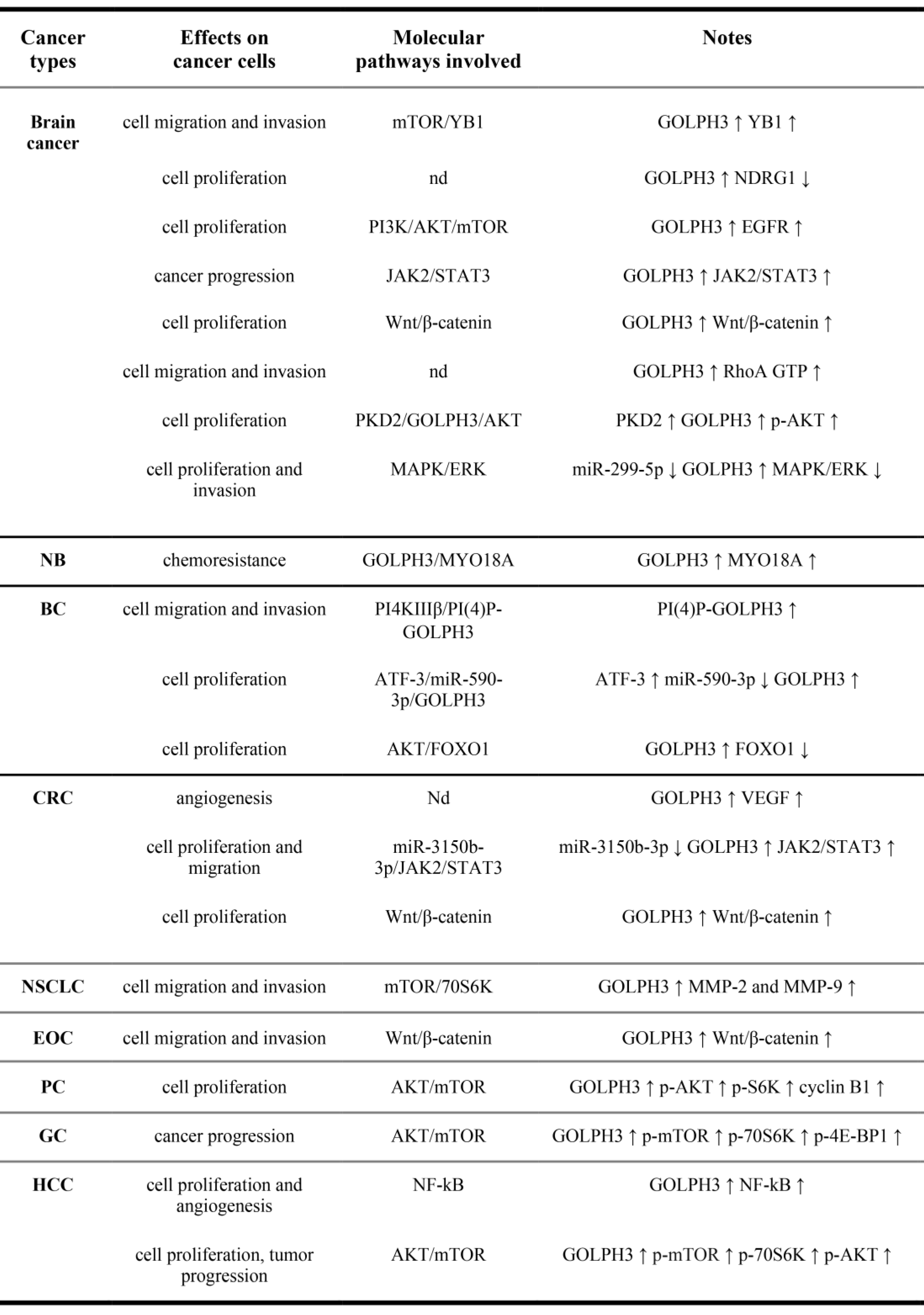 Figure 2. Effects of GOLPH3 up-regulation in solid tumors and molecular pathways involved. NB, Neuroblastoma; BC, Breast cancer; CRC, Colorectal cancer; NSCLC, Non-Small Cell Lung Cancer; EOC, Epithelial Ovarian cancer; PC, Prostate cancer; GC, Gastric Cancer; HCC, hepatocellular carcinoma. (↑),upregulation; (↓), downregulation. nd, not determined. (p-), phosphorylated.
Figure 2. Effects of GOLPH3 up-regulation in solid tumors and molecular pathways involved. NB, Neuroblastoma; BC, Breast cancer; CRC, Colorectal cancer; NSCLC, Non-Small Cell Lung Cancer; EOC, Epithelial Ovarian cancer; PC, Prostate cancer; GC, Gastric Cancer; HCC, hepatocellular carcinoma. (↑),upregulation; (↓), downregulation. nd, not determined. (p-), phosphorylated.
Tumors in which the role of GOLPH3 had been better elucidated include glioma, breast cancer and colorectal cancer. In gliomas, the role of GOLPH3 in cancer cell growth, migration and invasiveness have been partly elucidated. Glioma is the most common tumor of the brain and the role of GOLPH3 in its etiology dates back to 2011, when Li and collaborators showed a more than half of their patients affected by glioma were positive for the overexpression of either GOLPH3 protein or RNA, and that the amount of GOLPH3 expression in those patients was associated with the severity of the tumor[19]. Accordingly, also a relation between low GOLPH3 expression and significantly longer median overall survival has been demonstrated[15]. In breast cancer, it has been shown that GOLPH3 is significantly upregulated in both cell coltures and in patients, and that its upregulation significantly correlates with the advanced clinical stage of breast cancer, poorly differentiated tumors and worse prognosis[12][20]. Moreover, upregulated GOLPH3 also influences the migration/invasion capacity of these cells[13]. Finally, GOLPH3 level has been also associated with the clinical stage of the disease, lymph node metastasis and infiltration degree in colorectal cancer[21][22] and, at the same time, seems to confer resistance to some chemotherapeutic drugs, although these last data need further validation. The molecular pathways and the de-regulations occurring in glioma, breast and colon cancer pathogenesis are summarized in Figure 2 and referenced in[18].
References
- Ira Mellman; Yosef Yarden; Endocytosis and Cancer. Cold Spring Harbor Perspectives in Biology 2013, 5, a016949-a016949, 10.1101/cshperspect.a016949.
- Mark G. Waugh; The Great Escape: how phosphatidylinositol 4-kinases and PI4P promote vesicle exit from the Golgi (and drive cancer). Biochemical Journal 2019, 476, 2321-2346, 10.1042/bcj20180622.
- Kenneth L. Scott; Lynda Chin; Signaling from the Golgi: mechanisms and models for Golgi phosphoprotein 3-mediated oncogenesis.. Clinical Cancer Research 2010, 16, 2229-34, 10.1158/1078-0432.CCR-09-1695.
- Christine C. Wu; Randall S. Taylor; Diana R. Lane; Mark S. Ladinsky; Julie A. Weisz; Kathryn E. Howell; GMx33: A Novel Family of trans-Golgi Proteins Identified by Proteomics. Traffic 2000, 1, 963-975, 10.1111/j.1600-0854.2000.11206.x.
- Alexander W. Bell; Malcolm A. Ward; Hamzah N. M. Freeman; Jyoti S. Choudhary; Dipti Chotai; Ali Fazel; Jennifer N. Gushue; Jacques Paiement; Sandrine Palcy; Eric Chevet; et al.Roberto SolariDavid Y. ThomasAdele RowleyJohn J. M. BergeronWalter P. BlackstockAlan P. LewisMyriam Lafrenière-Roula Proteomics Characterization of Abundant Golgi Membrane Proteins. Journal of Biological Chemistry 2000, 276, 5152-5165, 10.1074/jbc.m006143200.
- Holly C. Dippold; Michelle M. Ng; Suzette E. Farber-Katz; Sun-Kyung Lee; Monica L. Kerr; Marshall C. Peterman; Ronald Sim; Patricia A. Wiharto; Kenneth A. Galbraith; Swetha Madhavarapu; et al.Greg J. FuchsTimo MeerlooMarilyn G. FarquharHuilin ZhouSeth J. Field GOLPH3 Bridges Phosphatidylinositol-4- Phosphate and Actomyosin to Stretch and Shape the Golgi to Promote Budding. Cell 2009, 139, 337-351, 10.1016/j.cell.2009.07.052.
- Christopher M. Snyder; Gonzalo A. Mardones; Mark S. Ladinsky; Kathryn E. Howell; GMx33 Associates with the Trans-Golgi Matrix in a Dynamic Manner and Sorts within Tubules Exiting the GolgiD⃞V⃞. Molecular Biology of the Cell 2006, 17, 511-524, 10.1091/mbc.E05-07-0682.
- Stefano Sechi; Gianni Colotti; Giorgio Belloni; Vincenzo Mattei; Anna Frappaolo; Grazia Daniela Raffa; Margaret T. Fuller; Maria Grazia Giansanti; GOLPH3 Is Essential for Contractile Ring Formation and Rab11 Localization to the Cleavage Site during Cytokinesis in Drosophila melanogaster. PLOS Genetics 2014, 10, e1004305, 10.1371/journal.pgen.1004305.
- Kenneth L. Scott; Omar Kabbarah; Mei-Chih Liang; Elena Ivanova; Valsamo Anagnostou; Joyce Wu; Sabin Dhakal; Min Wu; Shujuan Chen; Tamar Feinberg; et al.Joseph HuangAbdel SaciHans R. WidlundDavid E. FisherYonghong XiaoDavid L. RimmAlexei ProtopopovKwok-Kin WongLynda Chin GOLPH3 modulates mTOR signalling and rapamycin sensitivity in cancer. Nature 2009, 459, 1085-90, 10.1038/nature08109.
- Stefano Sechi; Anna Frappaolo; Giorgio Belloni; Gianni Colotti; Maria Grazia Giansanti; The multiple cellular functions of the oncoprotein Golgi phosphoprotein 3. Oncotarget 2015, 6, 3493-3506, 10.18632/oncotarget.3051.
- Ramya S. Kuna; Seth J. Field; GOLPH3: a Golgi phosphatidylinositol(4)phosphate effector that directs vesicle trafficking and drives cancer. Journal of Lipid Research 2018, 60, 269-275, 10.1194/jlr.R088328.
- Zhaolei Zeng; Huanxin Lin; Xiaohui Zhao; Guanglin Liu; X. Wang; Ruihua Xu; Kun Chen; Jun Li; Libing Song; Overexpression of GOLPH3 Promotes Proliferation and Tumorigenicity in Breast Cancer via Suppression of the FOXO1 Transcription Factor. Clinical Cancer Research 2012, 18, 4059-4069, 10.1158/1078-0432.CCR-11-3156.
- E. Tokuda; T. Itoh; Junya Hasegawa; Takeshi Ijuin; Y. Takeuchi; Y. Irino; M. Fukumoto; T. Takenawa; Phosphatidylinositol 4-Phosphate in the Golgi Apparatus Regulates Cell-Cell Adhesion and Invasive Cell Migration in Human Breast Cancer. Cancer Research 2014, 74, 3054-3066, 10.1158/0008-5472.can-13-2441.
- Xu Zhang; Zhijun Ding; Jianbing Mo; Ben Sang; Qiong Shi; Jinxia Hu; Shao Xie; Wenjian Zhan; Ng Lu; Minglin Yang; et al.Wenbin BianXiuping ZhouRutong Yu GOLPH3 promotes glioblastoma cell migration and invasion via the mTOR-YB1 pathway in vitro. Molecular Carcinogenesis 2014, 54, 1252-1263, 10.1002/mc.22197.
- Jinxu Zhou; Tao Xu; Rong Qin; Yong Yan; Chao Chen; Yuanyuan Chen; Hongyu Yu; Chunyan Xia; Yicheng Lu; Xuehua Ding; et al.Yuhai WangXuejian CaiJuxiang Chen Overexpression of Golgi phosphoprotein-3 (GOLPH3) in glioblastoma multiforme is associated with worse prognosis. Journal of Neuro-Oncology 2012, 110, 195-203, 10.1007/s11060-012-0970-9.
- Stefanie L. Makowski; Thuy T.T. Tran; Seth J. Field; Emerging themes of regulation at the Golgi.. Current Opinion in Cell Biology 2017, 45, 17-23, 10.1016/j.ceb.2017.01.004.
- Riccardo Rizzo; Seetharaman Parashuraman; Giovanni D’Angelo; Alberto Luini; GOLPH3 and oncogenesis: What is the molecular link?. Tissue and Cell 2017, 49, 170-174, 10.1016/j.tice.2016.06.008.
- Stefano Sechi; Anna Frappaolo; Angela Karimpour-Ghahnavieh; Roberto Piergentili; Maria Grazia Giansanti; Oncogenic Roles of GOLPH3 in the Physiopathology of Cancer. International Journal of Molecular Sciences 2020, 21, 933, 10.3390/ijms21030933.
- Xue-Yuan Li; Wei Liu; Shuang-Feng Chen; Lian-Qun Zhang; Xin-Gang Li; Le-Xin Wang; Expression of the Golgi phosphoprotein-3 gene in human gliomas: a pilot study. Journal of Neuro-Oncology 2011, 105, 159-163, 10.1007/s11060-011-0573-x.
- Shicong Tang; Hong Pan; Wei Wei; Huawei Yang; Jianlun Liu; Rirong Yang; GOLPH3: a novel biomarker that correlates with poor survival and resistance to chemotherapy in breast cancer. Oncotarget 2017, 8, 105155-105169, 10.18632/oncotarget.21927.
- Yan-Ta Guo; Cheng-Zhi Qiu; Zhong-Xin Huang; Wai-Shi Yu; Xiao-Feng Yang; Ming-Zhen Wang; Correlational research of Golgi phosphorylation protein 3 expression in colorectal cancer. World Journal of Gastroenterology 2015, 21, 13473-13479, 10.3748/wjg.v21.i48.13473.
- Bo Zhou; Gongping Wang; Shegan Gao; Ye Chen; Canhui Jin; Zengfang Wang; Yantong Yang; Zhikun Ma; Wei Zhang; Xiaoshan Feng; et al. Expression of GOLPH3 protein in colon cancer tissues and its association with the prognosis of patients. Oncology Letters 2016, 12, 3936-3940, 10.3892/ol.2016.5215.

