Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Christine Alewine | -- | 2307 | 2022-09-08 03:28:53 | | | |
| 2 | Catherine Yang | Meta information modification | 2307 | 2022-09-08 03:30:54 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Skorupan, N.; Dominguez, M.P.; Ricci, S.L.; Alewine, C. Tumor Microenvironment of Pancreatic Ductal Adenocarcinoma. Encyclopedia. Available online: https://encyclopedia.pub/entry/26983 (accessed on 26 July 2026).
Skorupan N, Dominguez MP, Ricci SL, Alewine C. Tumor Microenvironment of Pancreatic Ductal Adenocarcinoma. Encyclopedia. Available at: https://encyclopedia.pub/entry/26983. Accessed July 26, 2026.
Skorupan, Nebojsa, Mayrel Palestino Dominguez, Samuel L. Ricci, Christine Alewine. "Tumor Microenvironment of Pancreatic Ductal Adenocarcinoma" Encyclopedia, https://encyclopedia.pub/entry/26983 (accessed July 26, 2026).
Skorupan, N., Dominguez, M.P., Ricci, S.L., & Alewine, C. (2022, September 08). Tumor Microenvironment of Pancreatic Ductal Adenocarcinoma. In Encyclopedia. https://encyclopedia.pub/entry/26983
Skorupan, Nebojsa, et al. "Tumor Microenvironment of Pancreatic Ductal Adenocarcinoma." Encyclopedia. Web. 08 September, 2022.
Copy Citation
Pancreatic cancer has a complex tumor microenvironment which engages in extensive crosstalk between cancer cells, cancer-associated fibroblasts, and immune cells. Many of these interactions contribute to tumor resistance to anti-cancer therapies. Using targeted drugs to disrupt interactions between these cells which can support cancer cell growth, invasion, and immune suppression has become an important area of exploration in the pancreatic cancer field. Pancreatic ductal adenocarcinoma (PDAC) is the most common histology of pancreatic cancer, representing >85% of all pancreatic cancer diagnoses.
immunotherapy
pancreatic cancer
stromal modifiers
1. Tumor Microenvironment in PDAC
The tumor microenvironment (TME) of PDAC contains numerous cell types and exhibits a resilient and exuberant desmoplastic reaction (Figure 1). Desmoplasia generates a dense ECM composed of collagen, fibronectin, laminin, and hyaluronic acid that is synthesized by cancer-associated fibroblasts (CAFs). The resulting fibrosis generates interstitial fluid pressures than can exceed mean arterial pressures. This precipitates vascular compression and creates a considerable barrier to therapeutics that must exit the circulation and reach the vicinity of cancer cells [1]. Several subtypes of CAFs with unique gene signatures and specialized functions have been identified: myofibroblastic CAFs (myCAFs), inflammatory CAFs (iCAFs), and antigen-presenting CAFs (apCAFs) [2][3][4]. Initially, it was thought that all CAF populations arose from pancreatic stellate cells (PSCs) and differentiated to the various subtypes under the influence of environmental cues within the TME, including cytokine and growth factor gradients. However, it has recently been demonstrated that PSC-derived CAFs give rise only to a minor population of myCAFs [3] and that alternative cell populations differentiate into apCAFs [5]. CAFs release cytokines, chemokines, adhesion molecules, and growth factors that influence immune and endothelial cells. CAFs also regulate ECM components that can contribute to metastatic spread and tumor aggressiveness [6][7].
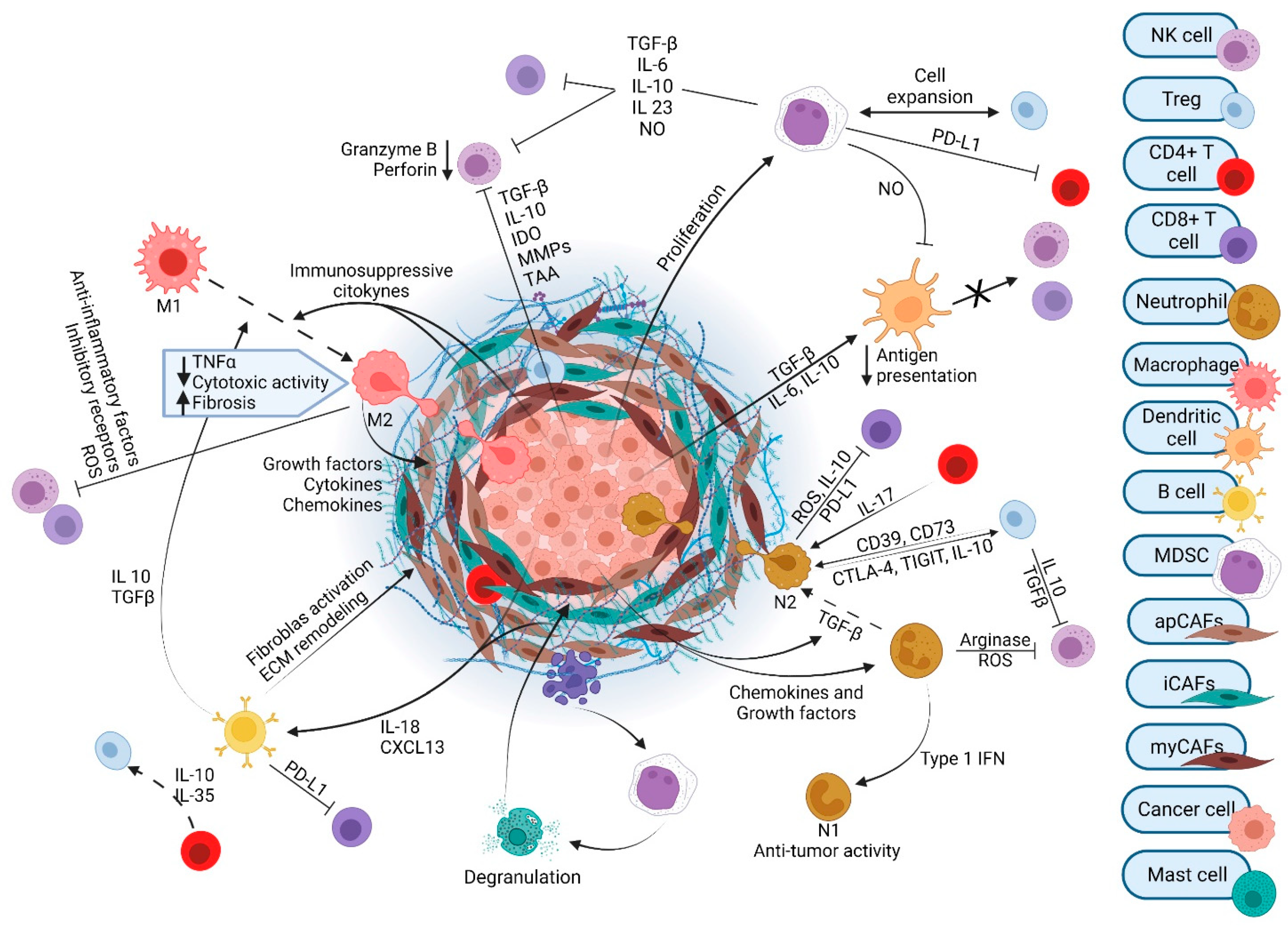
Figure 1. The tumor microenvironment of pancreatic ductal adenocarcinoma.
Amongst the cell types that differentiate into PDAC CAFs, the role of PSC-derived CAFs has been extensively studied and has revealed both a tumor-promoting and tumor-restraining role for these cells and their products [3][8][9]. The activation of PSCs results in their transformation from a quiescent to a myofibroblast-like phenotype that expresses high levels of alpha smooth muscle actin (αSMA). In patients with early-stage (T1–T2) PDAC, moderate-to-strong αSMA expression was associated with poorer clinical outcomes compared to tumors with lower levels of αSMA expression [10]. PSC-derived CAFs can inhibit cancer cell apoptosis and promote chemoresistance and disease recurrence. PSCs can accompany cancer cells to distant metastatic sites and help to establish a supportive niche for their growth [11]. In fact, evidence supports that survival of tumor cells in the inhospitable PDAC TME depends on CAFs. Acute destruction of FAP+ (fibroblast-expressing activation protein) CAFs caused rapid hypoxic necrosis of cancer and stromal cells that is interferon-γ (IFN-γ)- and tumor necrosis factor-α (TNF-α)-dependent [12]. Others have delineated a role for FAP(+)-CAFs in immunosuppression [13]. Activated PSCs secrete a dense ECM that modulates tumor stiffness and invasiveness through increased expression of the type IV collagenase matrix metalloproteinase-2 (MMP-2) [9]. Meflin, a glycosylphosphatidylinositol-anchored protein in CAFs, interacts with lysyl oxidase to inhibit collagen crosslinking activity and reduce tissue stiffness. The induced expression of meflin by both genetic and pharmacological approaches increased tumor vessel area in murine PDAC, improved drug delivery, and increased tumor chemosensitivity [14]. This is one of many pre-clinical studies showing that decreased ECM stiffness enhances therapeutic efficacy [1][15]. From these data, one might conclude that elimination of CAFs and reduction in ECM should inhibit PDAC and improve patient outcomes. However, negative clinical studies using Sonic Hedgehog (Hh) inhibitors to ablate CAFs in PDAC patients required the field to reconsider the role of this cell type in PDAC [16][17]. Subsequently, it was found that, paradoxically, depletion of type I collagen from PDAC-bearing mouse stroma significantly decreased animal survival [18]. Furthermore, ablation of αSMA(+) fibroblasts resulted in highly hypoxic, undifferentiated tumors with a more aggressive phenotype [19]. Similarly, inhibition of collagen crosslinking by LOXL2 increased PDAC growth and reduced overall survival [20]. These studies demonstrate that while there is clearly a sub-population of CAFs that facilitates cancer growth, some portion of the CAF population also appears to have a tumor-restraining role.
In the last few years, research into CAFs and their diverse sub-populations has provided increased insight. Nevertheless, correlating individual CAF subtypes with a universally bad (tumor-promoting) or good (tumor-restraining) phenotype is more difficult. The literature is generally concordant in condemning iCAFs as bad actors in the PDAC TME. These cells have been implicated in neoplastic progression to PDAC through induction of inflammation and complement regulatory factors [21]. Moreover, iCAFs can aid the preservation of cancer stem cells and facilitate chemotherapy resistance by modulating TME metabolism [22]. Conversely, myCAFs’ effect on cancer cells appears variable and situational. Depletion of myCAFs by Hh inhibition changes the balance of T cell subsets to generate a more immunosuppressive tumor, while at the same time impairing tumor growth, at least in the short-term setting [23]. It appears that apCAFs could fall on the good side in some contexts; these cells can induce CD25 and CD69 immune activators in co-cultured T cells [2]. However, there is evidence showing that apCAFs differentiated from mesothelial cells transform CD4+ T cells into Treg cells in mice [5]. Adding to the complexity, it has been suggested that iCAF and myCAF determination is not static, but that cells could be “interconvertible between types” under the influence of the correct chemical signals. In fact, treatment of tumors with inhibitors of the JAK/STAT pathway, a critical agent in iCAF differentiation, resulted in an increased myCAF to iCAF ratio [24]. Recently, a fourth type of CAF, highly activated metabolic state CAF (meCAF), was identified by single-cell analysis of PDAC specimens of varying desmoplastic exuberance. These meCAFs were highly prevalent in low density tumors and predictive of poorer prognosis. However, their presence also predicted better response to GN/anti-programmed cell death protein 1 (PD-1) blockade in PDAC patients, an effect attributed to enhanced immune surveillance compared to tumors with high desmoplasia [25]. The varying roles of CAFs make pharmacological targeting of CAFs and ECM components highly complex, since proposed strategies must selectively eliminate tumor-promoting elements without inhibiting or eradicating tumor-restraining ones. Cautious selection of targets after rigorous pre-clinical testing is necessary to offer patients the least chance of unintentional harm.
2. PDAC Is Defined by an Immunosuppressive TME
Mutations in the KRAS oncogene are present in >90% of PDAC patients and activation of this pathway defines the disease [26][27][28]. Mutated KRAS in combination with inflammation or loss of key tumor suppressors drives progression of pre-malignant lesions to PDAC and is implicated in the recruitment of an immunosuppressive cellular milieu through KRAS-driven production of pro-inflammatory cytokines and chemokines [29][30][31][32]. Oncogenic KRAS has also been implicated in tumor immune evasion [33][34]. The role of mutated KRAS in establishing the immunosuppressive PDAC TME has been well described by others [35][36][37].
PDAC tumors have robust infiltration by T cells. Unfortunately, most of these cells promote tumorigenesis; cytotoxic T cells are infrequent in the PDAC TME. The most abundant T cell subtype is CD4+ regulatory T (Treg) cells. Tregs play a crucial role in warding off the host immune system. Tregs are increased in PDAC, conferring poor prognosis in patients. Several depletion experiments established Tregs to be suppressors of anti-tumor immune responses [38]. Interestingly, most cytotoxic CD8+ T cells are excluded from the vicinity of pancreatic cancer cells. In patients, the spatial proximity of cytotoxic CD8+ T cells, but not CD4+ T cells or total T cells, to pancreatic cancer cells correlates with increased overall survival [39].
The majority of immune cells in the TME are of myeloid origin. These include tumor-associated macrophages (TAMs), granulocytes, and inflammatory monocytes. During pre-cancerous stages, activated KRAS actively recruits these cells to the TME. TAMs are some of the most abundant immune cells in PDAC and their multiple roles have been extensively described previously [40]. TAMs can generally be categorized as either M1 or M2 polarized. M1 macrophages are considered tumor suppressive. When activated, they secrete TNF-α, interleukin (IL)-12, IL-1α, and IFN-γ, which can have a tumoricidal effect. Conversely, M2 macrophages are generally immunosuppressive. Their products, such as transforming growth factor-beta (TGF-β) and IL-10, tend to be tumor-promoting [41]. TAMs do not operate in isolation. TAMs and collagen in the TME interact to shape each other. High collagen density, as found in PDAC, promotes an immunosuppressive macrophage phenotype [42]. At the same time, TAMs can internalize collagen matrix through the action of the mannose receptor (MRC1), resulting in increased arginine synthesis from biproducts of lysosomal collagen breakdown. The high levels of arginine result in increased production of reactive nitrogen species, which in turn promote a profibrotic phenotype in PSCs. This leads to increased fibrosis and formation of more collagen [43]. TAMs can also affect cancer cell programming. In fact, the presence of TNF-α-secreting macrophages can push cancer cells to take on a more aggressive basal-like subtype [44]. Additionally, circulating monocytes and TAMs contribute to the development of the pre-metastatic niche by activating resident hepatic stellate cells. This promotes a fibrotic microenvironment that sustains metastatic tumor growth [45]. TAMs serve many roles in the PDAC TME.
MDSCs (myeloid-derived suppressor cells) are also derived from myeloid cells. They can be subtyped into monocytic or granulocytic and are known to exert immunosuppressive effects on T cells via arginase, nitric oxide synthase, TGF-β, IL-10, and COX2. MDSCs are recruited early in the process of carcinogenesis and promote the formation and maintenance of pre-neoplastic lesions. MDSCs also recruit Tregs to the TME. A more precise understanding of MDSCs has been difficult to achieve given their heterogeneity in both mice and humans [46].
Neutrophils are essential infiltrating immune cells in the PDAC TME, but tumor-associated neutrophils (TANs) are less mechanistically established in pancreatic carcinogenesis as compared with TAMs. TANs are detected even at early stages of the PDAC development. In mice, knockout of CXCR2, the primary neutrophil chemotaxis receptor, inhibits TAN infiltration into tumors, leading to T cell-dependent tumor growth inhibition [47]. Some studies classify TANs into two polarization states, tumor-suppressing N1 neutrophils and tumor-promoting N2 neutrophils [48]. Pro-inflammatory or immunostimulatory cytokines, such as IL-12, CXCL9, CXCL10, and CCL3, are released from N1 neutrophils and facilitate recruitment and activation of CD8+ T cells. On the other hand, exposure to TGF-β transforms neutrophils to the N2 phenotype [48]. N2 neutrophils have been reported to have strong immunosuppressive and tumor-supporting functions, including the promotion of tumor metastases and angiogenesis. Poor patient outcomes are associated with high intratumoral neutrophils in advanced cancer patients [49].
Dendritic cells (DCs) are professional antigen-presenting cells (APCs) that participate in both innate and adaptive immune responses and are critical to boosting immune responses to antigens, including tumor-associated antigens. DC responses are impaired in patients with PDAC. Specifically, PDAC secretes cytokines such as IL-6, IL-10, and TGF-β, which reduce the stimulatory capacity of DCs [50]. Overall numbers of circulating DCs are also noted to be lower in PDAC patients [51][52]. Interestingly, function and numbers of circulating DCs rebound following surgical resection of tumors. Within PDAC tumors, conventional DCs (cDCs) are largely excluded, a process which appears to begin in pre-malignant pancreas lesions [53][54]. In mice, restoration of cDC populations to established tumors is alone insufficient to break immune tolerance and regress tumors; stimulation to overcome low DC function is also required, including the presence of tumor antigens [54]. Despite this, increased numbers of circulating DCs and tumor DCs have been associated with better survival in patients with pancreatic cancer [55][56].
The role of B cells in PDAC tumorigenesis remains controversial. While B cells clustered in tertiary lymphoid structures are associated with better outcome in PDAC patients, manipulations that increase B cells have negative impacts on survival in several PDAC mouse models [57]. Most B cells are considered pro-inflammatory and immune-stimulatory, while ~10% are immunosuppressive. Under hypoxic conditions, CXCL13 secreted by CAFs attracts immunosuppressive B cells to tumors [58], which can then induce M2 polarization of TAMs [59]. In addition, B cells can activate CAFs via the soluble factor platelet derived growth factor-B (PDGF-B) [60][61]. It is difficult to reconcile the pro-tumor role of B cells in mice with developing tumors with the human data from well-established tumors. This apparent paradox suggests that B cells play different roles as the tumor progresses and evolves.
Natural killer (NK) cells are defined by the lack of surface T cell receptors (TCRs), the expression of the neural cell adhesion molecule (NCAM), and the natural cytotoxicity receptor (NCR) NKp46. NK cells recognize and directly kill virus-infected or tumor cells without prior antigen stimulation [62]. NK cells can kill by multiple mechanisms, including secretion of perforin to destroy cell membranes, release of granzymes for a lytic killing effect, or activation of the Fas/FasL pathway to induce apoptosis of target cells. Activated NK cells also secrete cytokines, such as TNF-α, IFN-γ, and granulocyte-macrophage colony-stimulating factor (GM-CSF), which trigger activation and recruitment of other innate and adaptive immune cells that broaden and strengthen the anti-tumor immune response [63]. For example, IFN-γ secretion by NK cells is critical in shaping T cell responses, including TH1 polarization and CD8+ T cell activation [64]. PDAC patients have normal numbers of peripheral NK cells, but NK-cell activity is progressively impaired at more advanced stages of disease. NK activating receptors NKG2D and NKp30 are expressed at lower levels in PDAC patients [65]. In addition, these cells have decreased cytotoxic activity, low IFN-γ expression, and high intracellular levels of IL-10. Within tumor tissue, NK cells are largely excluded and display a decreased activity and toxicity potential [66]. In PDAC, several mechanisms impair the NK cell function and polarize NK cells towards a tumor-promoting phenotype. Cancer cells suppress NK cell function through expression of TGF-β, IL-10, indoleamine 2,3-dioxygenase (IDO), and matrix metalloproteinases (MMPs), which impair NK cell tumor recognition and killing via the downregulation of cytotoxicity receptors. Another mechanism of NK-cell inhibition is the secretion of the Igγ-1 chain C region (IGHG1), which competitively binds to the Fcγ receptor of NK cells, reducing antibody-dependent cell-mediated cytotoxicity (ADCC). Both cancer cells and PSC-derived CAFs secrete IL-18, IL-10, and TGF-β, all of which diminish NK-cell function [62]. Interestingly, chemotherapy can restore NK-cell-mediated anti-tumor activity of endogenous NK cells in mouse models, an intriguing therapeutic side effect in the age of immunotherapy [67].
References
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429.
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123.
- Helms, E.J.; Berry, M.W.; Chaw, R.C.; DuFort, C.C.; Sun, D.; Onate, M.K.; Oon, C.; Bhattacharyya, S.; Sanford-Crane, H.; Horton, W.; et al. Mesenchymal Lineage Heterogeneity Underlies Nonredundant Functions of Pancreatic Cancer-Associated Fibroblasts. Cancer Discov. 2022, 12, 484–501.
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596.
- Huang, H.; Wang, Z.; Zhang, Y.; Pradhan, R.N.; Ganguly, D.; Chandra, R.; Murimwa, G.; Wright, S.; Gu, X.; Maddipati, R.; et al. Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell 2022, 40, 656–673.e657.
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926.
- Vennin, C.; Melenec, P.; Rouet, R.; Nobis, M.; Cazet, A.S.; Murphy, K.J.; Herrmann, D.; Reed, D.A.; Lucas, M.C.; Warren, S.C.; et al. CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nat. Commun. 2019, 10, 3637.
- Vonlaufen, A.; Joshi, S.; Qu, C.; Phillips, P.A.; Xu, Z.; Parker, N.R.; Toi, C.S.; Pirola, R.C.; Wilson, J.S.; Goldstein, D.; et al. Pancreatic stellate cells: Partners in crime with pancreatic cancer cells. Cancer Res. 2008, 68, 2085–2093.
- Schneiderhan, W.; Diaz, F.; Fundel, M.; Zhou, S.; Siech, M.; Hasel, C.; Moller, P.; Gschwend, J.E.; Seufferlein, T.; Gress, T.; et al. Pancreatic stellate cells are an important source of MMP-2 in human pancreatic cancer and accelerate tumor progression in a murine xenograft model and CAM assay. J. Cell Sci. 2007, 120, 512–519.
- Wang, L.M.; Silva, M.A.; D’Costa, Z.; Bockelmann, R.; Soonawalla, Z.; Liu, S.; O’Neill, E.; Mukherjee, S.; McKenna, W.G.; Muschel, R.; et al. The prognostic role of desmoplastic stroma in pancreatic ductal adenocarcinoma. Oncotarget 2016, 7, 4183–4194.
- Xu, Z.; Vonlaufen, A.; Phillips, P.A.; Fiala-Beer, E.; Zhang, X.; Yang, L.; Biankin, A.V.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; et al. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am. J. Pathol. 2010, 177, 2585–2596.
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 2010, 330, 827–830.
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217.
- Iida, T.; Mizutani, Y.; Esaki, N.; Ponik, S.M.; Burkel, B.M.; Weng, L.; Kuwata, K.; Masamune, A.; Ishihara, S.; Haga, H.; et al. Pharmacologic conversion of cancer-associated fibroblasts from a protumor phenotype to an antitumor phenotype improves the sensitivity of pancreatic cancer to chemotherapeutics. Oncogene 2022, 41, 2764–2777.
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013, 4, 2516.
- Catenacci, D.V.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 4284–4292.
- Ko, A.H.; LoConte, N.; Tempero, M.A.; Walker, E.J.; Kate Kelley, R.; Lewis, S.; Chang, W.C.; Kantoff, E.; Vannier, M.W.; Catenacci, D.V.; et al. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas 2016, 45, 370–375.
- Chen, Y.; Kim, J.; Yang, S.; Wang, H.; Wu, C.J.; Sugimoto, H.; LeBleu, V.S.; Kalluri, R. Type I collagen deletion in alphaSMA(+) myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 2021, 39, 548–565.e546.
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734.
- Jiang, H.; Torphy, R.J.; Steiger, K.; Hongo, H.; Ritchie, A.J.; Kriegsmann, M.; Horst, D.; Umetsu, S.E.; Joseph, N.M.; McGregor, K.; et al. Pancreatic ductal adenocarcinoma progression is restrained by stromal matrix. J. Clin. Investig 2020, 130, 4704–4709.
- Gieniec, K.A.; Butler, L.M.; Worthley, D.L.; Woods, S.L. Cancer-associated fibroblasts-heroes or villains? Br. J. Cancer 2019, 121, 293–302.
- Hu, B.; Wu, C.; Mao, H.; Gu, H.; Dong, H.; Yan, J.; Qi, Z.; Yuan, L.; Dong, Q.; Long, J. Subpopulations of cancer-associated fibroblasts link the prognosis and metabolic features of pancreatic ductal adenocarcinoma. Ann. Transl. Med. 2022, 10, 262.
- Steele, N.G.; Biffi, G.; Kemp, S.B.; Zhang, Y.; Drouillard, D.; Syu, L.; Hao, Y.; Oni, T.E.; Brosnan, E.; Elyada, E.; et al. Inhibition of Hedgehog Signaling Alters Fibroblast Composition in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 2023–2037.
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301.
- Wang, Y.; Liang, Y.; Xu, H.; Zhang, X.; Mao, T.; Cui, J.; Yao, J.; Wang, Y.; Jiao, F.; Xiao, X.; et al. Single-cell analysis of pancreatic ductal adenocarcinoma identifies a novel fibroblast subtype associated with poor prognosis but better immunotherapy response. Cell Discov. 2021, 7, 36.
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178.
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52.
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744.
- Cheng, H.; Fan, K.; Luo, G.; Fan, Z.; Yang, C.; Huang, Q.; Jin, K.; Xu, J.; Yu, X.; Liu, C. Kras(G12D) mutation contributes to regulatory T cell conversion through activation of the MEK/ERK pathway in pancreatic cancer. Cancer Lett. 2019, 446, 103–111.
- Bansod, S.; Dodhiawala, P.B.; Lim, K.H. Oncogenic KRAS-Induced Feedback Inflammatory Signaling in Pancreatic Cancer: An Overview and New Therapeutic Opportunities. Cancers 2021, 13, 5481.
- Liou, G.Y.; Döppler, H.; Necela, B.; Edenfield, B.; Zhang, L.; Dawson, D.W.; Storz, P. Mutant KRAS-induced expression of ICAM-1 in pancreatic acinar cells causes attraction of macrophages to expedite the formation of precancerous lesions. Cancer Discov. 2015, 5, 52–63.
- Pylayeva-Gupta, Y.; Lee, K.E.; Hajdu, C.H.; Miller, G.; Bar-Sagi, D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 2012, 21, 836–847.
- Ischenko, I.; D’Amico, S.; Rao, M.; Li, J.; Hayman, M.J.; Powers, S.; Petrenko, O.; Reich, N.C. KRAS drives immune evasion in a genetic model of pancreatic cancer. Nat. Commun. 2021, 12, 1482.
- Muthalagu, N.; Monteverde, T.; Raffo-Iraolagoitia, X.; Wiesheu, R.; Whyte, D.; Hedley, A.; Laing, S.; Kruspig, B.; Upstill-Goddard, R.; Shaw, R.; et al. Repression of the Type I Interferon Pathway Underlies MYC- and KRAS-Dependent Evasion of NK and B Cells in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2020, 10, 872–887.
- Javadrashid, D.; Baghbanzadeh, A.; Derakhshani, A.; Leone, P.; Silvestris, N.; Racanelli, V.; Solimando, A.G.; Baradaran, B. Pancreatic Cancer Signaling Pathways, Genetic Alterations, and Tumor Microenvironment: The Barriers Affecting the Method of Treatment. Biomedicines 2021, 9, 373.
- Gu, M.; Gao, Y.; Chang, P. KRAS Mutation Dictates the Cancer Immune Environment in Pancreatic Ductal Adenocarcinoma and Other Adenocarcinomas. Cancers 2021, 13, 2429.
- Hamarsheh, S.A.; Groß, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 5439.
- Joshi, N.S.; Akama-Garren, E.H.; Lu, Y.; Lee, D.Y.; Chang, G.P.; Li, A.; DuPage, M.; Tammela, T.; Kerper, N.R.; Farago, A.F.; et al. Regulatory T Cells in Tumor-Associated Tertiary Lymphoid Structures Suppress Anti-tumor T Cell Responses. Immunity 2015, 43, 579–590.
- Ajina, R.; Weiner, L.M. T cell Immunity in Pancreatic Cancer. Pancreas 2020, 49, 1014–1023.
- Poh, A.R.; Ernst, M. Tumor-Associated Macrophages in Pancreatic Ductal Adenocarcinoma: Therapeutic Opportunities and Clinical Challenges. Cancers 2021, 13, 2860.
- Stott, M.C.; Oldfield, L.; Hale, J.; Costello, E.; Halloran, C.M. Recent advances in understanding pancreatic cancer. Fac. Rev. 2022, 11, 9.
- Larsen, A.M.H.; Kuczek, D.E.; Kalvisa, A.; Siersbæk, M.S.; Thorseth, M.L.; Johansen, A.Z.; Carretta, M.; Grøntved, L.; Vang, O.; Madsen, D.H. Collagen Density Modulates the Immunosuppressive Functions of Macrophages. J. Immunol. 2020, 205, 1461–1472.
- LaRue, M.M.; Parker, S.; Puccini, J.; Cammer, M.; Kimmelman, A.C.; Bar-Sagi, D. Metabolic reprogramming of tumor-associated macrophages by collagen turnover promotes fibrosis in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2022, 119, e2119168119.
- Tu, M.; Klein, L.; Espinet, E.; Georgomanolis, T.; Wegwitz, F.; Li, X.; Urbach, L.; Danieli-Mackay, A.; Kuffer, S.; Bojarczuk, K.; et al. TNF-alpha-producing macrophages determine subtype identity and prognosis via AP1 enhancer reprogramming in pancreatic cancer. Nat. Cancer 2021, 2, 1185–1203.
- Nielsen, S.R.; Quaranta, V.; Linford, A.; Emeagi, P.; Rainer, C.; Santos, A.; Ireland, L.; Sakai, T.; Sakai, K.; Kim, Y.S.; et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat. Cell Biol. 2016, 18, 549–560.
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174.
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—Clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540.
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194.
- Jin, L.; Kim, H.S.; Shi, J. Neutrophil in the Pancreatic Tumor Microenvironment. Biomolecules 2021, 11, 1170.
- Bellone, G.; Carbone, A.; Smirne, C.; Scirelli, T.; Buffolino, A.; Novarino, A.; Stacchini, A.; Bertetto, O.; Palestro, G.; Sorio, C.; et al. Cooperative induction of a tolerogenic dendritic cell phenotype by cytokines secreted by pancreatic carcinoma cells. J. Immunol. 2006, 177, 3448–3460.
- Takahashi, K.; Toyokawa, H.; Takai, S.; Satoi, S.; Yanagimoto, H.; Terakawa, N.; Araki, H.; Kwon, A.H.; Kamiyama, Y. Surgical influence of pancreatectomy on the function and count of circulating dendritic cells in patients with pancreatic cancer. Cancer Immunol. Immunother. CII 2006, 55, 775–784.
- Tjomsland, V.; Sandström, P.; Spångeus, A.; Messmer, D.; Emilsson, J.; Falkmer, U.; Falkmer, S.; Magnusson, K.E.; Borch, K.; Larsson, M. Pancreatic adenocarcinoma exerts systemic effects on the peripheral blood myeloid and plasmacytoid dendritic cells: An indicator of disease severity? BMC Cancer 2010, 10, 87.
- Dallal, R.M.; Christakos, P.; Lee, K.; Egawa, S.; Son, Y.I.; Lotze, M.T. Paucity of dendritic cells in pancreatic cancer. Surgery 2002, 131, 135–138.
- Hegde, S.; Krisnawan, V.E.; Herzog, B.H.; Zuo, C.; Breden, M.A.; Knolhoff, B.L.; Hogg, G.D.; Tang, J.P.; Baer, J.M.; Mpoy, C.; et al. Dendritic Cell Paucity Leads to Dysfunctional Immune Surveillance in Pancreatic Cancer. Cancer Cell 2020, 37, 289–307.e289.
- Yamamoto, T.; Yanagimoto, H.; Satoi, S.; Toyokawa, H.; Yamao, J.; Kim, S.; Terakawa, N.; Takahashi, K.; Kwon, A.H. Circulating myeloid dendritic cells as prognostic factors in patients with pancreatic cancer who have undergone surgical resection. J. Surg. Res. 2012, 173, 299–308.
- Deicher, A.; Andersson, R.; Tingstedt, B.; Lindell, G.; Bauden, M.; Ansari, D. Targeting dendritic cells in pancreatic ductal adenocarcinoma. Cancer Cell Int. 2018, 18, 85.
- Castino, G.F.; Cortese, N.; Capretti, G.; Serio, S.; Di Caro, G.; Mineri, R.; Magrini, E.; Grizzi, F.; Cappello, P.; Novelli, F.; et al. Spatial distribution of B cells predicts prognosis in human pancreatic adenocarcinoma. Oncoimmunology 2016, 5, e1085147.
- Lee, K.E.; Spata, M.; Bayne, L.J.; Buza, E.L.; Durham, A.C.; Allman, D.; Vonderheide, R.H.; Simon, M.C. Hif1a Deletion Reveals Pro-Neoplastic Function of B Cells in Pancreatic Neoplasia. Cancer Discov. 2016, 6, 256–269.
- Delvecchio, F.R.; Goulart, M.R.; Fincham, R.E.A.; Bombadieri, M.; Kocher, H.M. B cells in pancreatic cancer stroma. World J. Gastroenterol. 2022, 28, 1088–1101.
- Minici, C.; Testoni, S.; Della-Torre, E. B-Lymphocytes in the Pathophysiology of Pancreatic Adenocarcinoma. Front. Immunol. 2022, 13, 867902.
- Minici, C.; Rigamonti, E.; Lanzillotta, M.; Monno, A.; Rovati, L.; Maehara, T.; Kaneko, N.; Deshpande, V.; Protti, M.P.; De Monte, L.; et al. B lymphocytes contribute to stromal reaction in pancreatic ductal adenocarcinoma. Oncoimmunology 2020, 9, 1794359.
- Huber, M.; Brehm, C.U.; Gress, T.M.; Buchholz, M.; Alashkar Alhamwe, B.; von Strandmann, E.P.; Slater, E.P.; Bartsch, J.W.; Bauer, C.; Lauth, M. The Immune Microenvironment in Pancreatic Cancer. Int. J. Mol. Sci. 2020, 21, 7307.
- Van Audenaerde, J.R.M.; Roeyen, G.; Darcy, P.K.; Kershaw, M.H.; Peeters, M.; Smits, E.L.J. Natural killer cells and their therapeutic role in pancreatic cancer: A systematic review. Pharmacol. Ther. 2018, 189, 31–44.
- Wang, F.; Lau, J.K.C.; Yu, J. The role of natural killer cell in gastrointestinal cancer: Killer or helper. Oncogene 2021, 40, 717–730.
- Lee, H.S.; Leem, G.; Kang, H.; Jo, J.H.; Chung, M.J.; Jang, S.J.; Yoon, D.H.; Park, J.Y.; Park, S.W.; Song, S.Y.; et al. Peripheral natural killer cell activity is associated with poor clinical outcomes in pancreatic ductal adenocarcinoma. J. Gastroenterol. Hepatol. 2021, 36, 516–522.
- Marcon, F.; Zuo, J.; Pearce, H.; Nicol, S.; Margielewska-Davies, S.; Farhat, M.; Mahon, B.; Middleton, G.; Brown, R.; Roberts, K.J.; et al. NK cells in pancreatic cancer demonstrate impaired cytotoxicity and a regulatory IL-10 phenotype. Oncoimmunology 2020, 9, 1845424.
- Childs, R.W.; Berg, M. Bringing natural killer cells to the clinic: Ex vivo manipulation. Hematol. Am. Soc. Hematol. Educ. Program 2013, 2013, 234–246.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
08 Sep 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No