 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mohd Faiz Mustaffa | -- | 5120 | 2022-08-26 02:26:30 | | | |
| 2 | Beatrix Zheng | + 19 word(s) | 5139 | 2022-08-26 09:56:55 | | | | |
| 3 | Beatrix Zheng | Meta information modification | 5139 | 2022-08-26 09:58:16 | | |
Video Upload Options
The oral route is the most common and practical means of drug administration, particularly from a patient’s perspective. However, the pharmacokinetic profile of oral drugs depends on the rate of drug absorption through the intestinal wall before entering the systemic circulation. However, the enteric epithelium represents one of the major limiting steps for drug absorption, due to the presence of efflux transporters on the intestinal membrane, mucous layer, enzymatic degradation, and the existence of tight junctions along the intestinal linings. These challenges are more noticeable for hydrophilic drugs, high molecular weight drugs, and drugs that are substrates of the efflux transporters. Another challenge faced by oral drug delivery is the presence of first-pass hepatic metabolism that can result in reduced drug bioavailability. Over the years, a wide range of compounds have been investigated for their permeation-enhancing effect in order to circumvent these challenges. There is also a growing interest in developing nanocarrier-based formulation strategies to enhance the drug absorption.
1. Current Absorption Enhancers and Their Absorption-Enhancing Mechanisms to Improve the Pharmacokinetic Profile
1.1. Solubilizing Agents
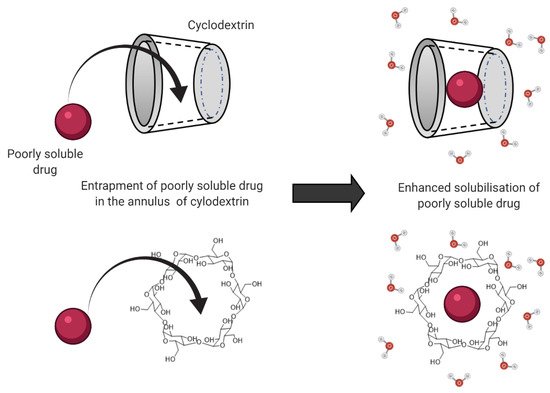
1.2. Bile Salts
| Drug (s) | Absorption Enhancer | Model | Results | Ref. |
|---|---|---|---|---|
| 5(6)-carboxyfluorescein | Sodium glycocholate (SGC) and sodium taurodeoxycholate (STDC) | In vitro: Caco-2 cell | SGC was a slightly better absorption enhancer for the 5(6)-carboxyfluorescein than STDC but not significant (p > 0.05). | [21] |
| Cefquinome | Sodium taurocholate | In vitro: Caco-2 cell | At 2 mmol/L sodium taurocholate, the transportation of cefquinome substantially increased. | [20] |
| In vivo: rat intestine | At 10 and 20 mmol/L sodium taurocholate, the absorption of the drug increased in a concentration-dependent manner. | |||
| Berberine chloride | Sodium deoxycholate | In vivo: rat intestine | AUC0–36h: 35.3-fold increase | [18] |
| Gliclazide | Taurocholic acid | In vivo: rat intestine | The microcapsules containing taurocholic acid increased the gliclazide absorption (p < 0.01). | [19] |
| EGFR2R-lytic hybrid peptide | Sodium taurodeoxycholate | In vitro: Caco-2 cell | Papp: 5.0-fold increase | [22] |
1.3. Chitosan
| Drug (s) | Absorption Enhancer | Model | Results | Ref. |
|---|---|---|---|---|
| Acyclovir | Chitosan | In vitro: Caco-2 cell | Papp: 124- and 143-fold increase | [31] |
| In vivo: rat intestine | AUC0–12 and AUC0–∞: 0.70- and 0.74-fold decrease Cmax: 0.56- and 0.63-fold decrease Tmax: 1.25- and 1.50-fold increase |
|||
| In vitro: Ussing chamber | Papp: 1.08- and 2.33-fold increase | |||
| Glucosamine hydrochloride | Chitosan | In vitro: Caco-2 cell | Papp: 1.9, 2.5 and 4.0-fold increase | [36] |
| In vivo: rat intestine | Cmax: 2.8-fold increase Tmax: no change AUC0−∞: 2.5-fold increase |
|||
| Salvianolic acid B | Chitosan | In vitro: Caco-2 cell | Papp: 4.43-fold increase | [27] |
| In vivo: rat intestine | AUC0–∞: 4.25-fold increase | |||
| Berberine | Chitosan hydrochloride | In vivo: rat intestine | AUC0–36: no improvement Cmax: no improvement |
[34] |
| Chitosan | In vivo: rat intestine | AUC0–36: maximum 2.5-fold increase | ||
| Amphotericin B | Trimethyl chitosan | In vitro: Caco-2 cell | Papp: 1.11-fold increase | [35] |
2. Formulation Strategies to Improve Pharmacokinetics Profile
2.1. Solid Lipid Nanoparticles (SLN)
2.2. Dimers
| Drug (s) | Model | Results | Ref. |
|---|---|---|---|
| 5(6)-carboxyfluorescein (CF), fluorescein isothiocyanate-labeled dextrans (FD4, FD10) and alendronate | In vitro: diffusion chamber | Papp: increased except for FD10. | [45] |
| In vivo: rat intestine | The greatest AUC achieved in the presence of Ac50-G2 (0.5%, w/v). | ||
| Camptothecin | In vivo: rat intestine | AUC: 2- to 3-fold increase Cmax: increased Tmax: no change |
[46] |
| Simvastatin | In vivo: rat intestine | AUC: increased Cmax: increased Tmax: 1.5-fold increase |
[47] |
| In vitro: Caco-2 cell | Papp: increased | ||
| Propranolol | In vitro Release Study (dialysis sac) | Papp: increased | [48] |
| In vitro: Caco-2 cell | AUC: increased | [49] |
2.3. Nanoemulsions
| Drug (s) | Model | Results | Ref. |
|---|---|---|---|
| Paeonol | In situ: single-pass intestine perfusion | Papp: 1.64-fold increase Ka: 0.65-fold increase |
[62] |
| In vitro: everted gut sacs | Papp: increased (p < 0.01) | ||
| In vitro: Caco-2 cell | Papp: increased | ||
| In vivo: rat intestinal uptake | AUC0→t: 4.27-fold increase Cmax: 4.02-fold increase Tmax: 40-min increase |
||
| Berberine hydrochloride | In vivo: rat intestinal uptake | AUC: 4.4-fold increase Cmax: 1.6-fold increase Tmax: 4.3-fold increase |
[61] |
| In vitro: Caco-2 cell | Papp: increased to 0.574 ± 0.18 × 10−8 cm/s | ||
| Curcumin | In vitro: Caco-2 cell | The digested nanoemulsion had the highest permeation rate (7.07 × 105 cm/s) | [56] |
| Candesartan cilexetil | In situ single-pass intestinal perfusion | Cellular uptake: 1.75-, 1.93-, and 1.84-fold increase in the duodenum, jejunum, and ileum, respectively. | [58] |
| In vitro: Caco-2 cell | The cellular uptake of CCN at 4 °C reduced 92% compared with that at 37 °C (p < 0.01) | ||
| In vivo: rat intestinal uptake | AUC: 10-fold increase Cmax: 27-fold increase Tmax: no change |
||
| Ibuprofen | In vitro diffusion chamber: rat intestinal membrane | Papp: 10.6-fold | [57] |
| In vivo: rat intestinal uptake | AUC 0–6h: 2.2-fold increase Cmax: 27-fold increase Tmax: no change |
References
- Savla, R.; Browne, J.; Plassat, V.; Wasan, K.M.; Wasan, E.K. Review and analysis of FDA approved drugs using lipid-based formulations. Drug Dev. Ind. Pharm. 2017, 43, 1743–1758.
- Sánchez-Navarro, M.; Garcia, J.; Giralt, E.; Teixidó, M. Using peptides to increase transport across the intestinal barrier. Adv. Drug Deliv. Rev. 2016, 106, 355–366.
- Gupta, S.; Kesarla, R.; Omri, A. Formulation Strategies to Improve the Bioavailability of Poorly Absorbed Drugs with Special Emphasis on Self-Emulsifying Systems. ISRN Pharm. 2013, 2013, 848043.
- Maher, S.; Heade, J.; McCartney, F.; Waters, S.; Bleiel, S.B.; Brayden, D. Effects of surfactant-based permeation enhancers on mannitol permeability, histology, and electrogenic ion transport responses in excised rat colonic mucosae. Int. J. Pharm. 2018, 539, 11–22.
- McCartney, F.; Rosa, M.; Brayden, D.J. Evaluation of Sucrose Laurate as an Intestinal Permeation Enhancer for Macromolecules: Ex Vivo and In Vivo Studies. Pharmaceutics 2019, 11, 565.
- Maroni, A.; Zema, L.; Del Curto, M.D.; Foppoli, A.; Gazzaniga, A. Oral colon delivery of insulin with the aid of functional adjuvants. Adv. Drug Deliv. Rev. 2012, 64, 540–556.
- Devasari, N.; Dora, C.P.; Singh, C.; Paidi, S.R.; Kumar, V.; Sobhia, M.E.; Suresh, S. Inclusion complex of erlotinib with sulfobutyl ether-β-cyclodextrin: Preparation, characterization, in silico, in vitro and in vivo evaluation. Carbohydr. Polym. 2015, 134, 547–556.
- Rubim, A.M.; Rubenick, J.B.; Maurer, M.; LaPorta, L.V.; Rolim, C.M.B. Inclusion complex of amiodarone hydrochloride with cyclodextrins: Preparation, characterization and dissolution rate evaluation. Braz. J. Pharm. Sci. 2017, 53.
- Rasheed, A. Cyclodextrins as Drug Carrier Molecule: A Review. Sci. Pharm. 2008, 76, 567–598.
- Wiedmann, T.S.; Kamel, L. Examination of the Solubilization of Drugs by Bile Salt Micelles. J. Pharm. Sci. 2002, 91, 1743–1764.
- Loftsson, T.; Moya-Ortega, M.D.; Alvarez-Lorenzo, C.; Concheiro, A. Pharmacokinetics of cyclodextrins and drugs after oral and parenteral administration of drug/cyclodextrin complexes. J. Pharm. Pharmacol. 2015, 68, 544–555.
- Pavlović, N.; Goločorbin-Kon, S.; Đanić, M.; Stanimirov, B.; Al-Salami, H.; Stankov, K.; Mikov, M. Bile Acids and Their Derivatives as Potential Modifiers of Drug Release and Pharmacokinetic Profiles. Front. Pharmacol. 2018, 9, 1283.
- Porter, C.; Trevaskis, N.; Charman, W. Lipids and lipid-based formulations: Optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov. 2007, 6, 231–248.
- Zughaid, H.; Forbes, B.; Martin, G.P.; Patel, N. Bile salt composition is secondary to bile salt concentration in determining hydrocortisone and progesterone solubility in intestinal mimetic fluids. Int. J. Pharm. 2012, 422, 295–301.
- Shaikh Permeability Enhancement Techniques for Poorly Permeable Drugs: A Review. J. Appl. Pharm. Sci. 2012, 2, 34–39.
- Kotze, A.; Luessen, H.; Thanou, M.; Verhoef, J.; De Boer, A.; Junginger, H. Chitosan and Chitosan Derivatives as Absorption Enhancers for Peptide Drugs Across Mucosal Epithelia. Drugs Pharm. Sci. 1999, 98, 341–386.
- Borchard, G.; Lueβen, H.L.; de Boer, A.G.; Verhoef, J.; Lehr, C.-M.; Junginger, H.E. The potential of mucoadhesive polymers in enhancing intestinal peptide drug absorption. III: Effects of chitosan-glutamate and carbomer on epithelial tight junctions in vitro. J. Control. Release 1996, 39, 131–138.
- Fan, D.; Wu, X.; Dong, W.; Sun, W.; Li, J.; Tang, X. Enhancement by sodium caprate and sodium deoxycholate of the gastrointestinal absorption of berberine chloride in rats. Drug Dev. Ind. Pharm. 2012, 39, 1447–1456.
- Mathavan, S.; Chen-Tan, N.; Arfuso, F.; Al-Salami, H. A comprehensive study of novel microcapsules incorporating gliclazide and a permeation enhancing bile acid: Hypoglycemic effect in an animal model of Type-1 diabetes. Drug Deliv. 2015, 23, 2869–2880.
- Yu, Q.; Cao, Y.; Liu, L.-F.; Jiang, S.-X.; Yang, Q. Enhancement of sodium taurocholate to the absorption of cefquinome. Pak. J. Pharm. Sci. 2016, 29, 139–143.
- Handali, S.; Moghimipour, E.; Tabassi, S.A.S.; Ramezani, M.; Lobenberg, R. Brush border membrane vesicle and Caco-2 cell line: Two experimental models for evaluation of absorption enhancing effects of saponins, bile salts, and some synthetic surfactants. J. Adv. Pharm. Technol. Res. 2016, 7, 75–79.
- Gaowa, A.; Horibe, T.; Kohno, M.; Kawakami, K. Bile Acid as an Effective Absorption Enhancer for Oral Delivery of Epidermal Growth Factor Receptor–Targeted Hybrid Peptide. J. Pharm. Sci. 2018, 107, 1322–1329.
- Tajdini, F.; Amini, M.A.; Nafissi-Varcheh, N.; Faramarzi, M.A. Production, physiochemical and antimicrobial properties of fungal chitosan from Rhizomucor miehei and Mucor racemosus. Int. J. Biol. Macromol. 2010, 47, 180–183.
- He, Z.G.; Lian, H.; Sun, J.; Yu, Y.P.; Liu, Y.H.; Wang, Y.J. Supramolecular micellar nanoaggregates based on a novel chitosan/vitamin E succinate copolymer for paclitaxel selective delivery. Int. J. Nanomed. 2011, 6, 3323–3334.
- Nadai, M.; Tajiri, C.; Yoshizumi, H.; Suzuki, Y.; Zhao, Y.L.; Kimura, M.; Tsunekawa, Y.; Hasegawa, T. Effect of Chitosan on Gastrointestinal Absorption of Water-Insoluble Drugs Following Oral Administration in Rats. Biol. Pharm. Bull. 2006, 29, 1941–1946.
- Dodane, V. Effect of chitosan on epithelial permeability and structure. Int. J. Pharm. 1999, 182, 21–32.
- Liang, K.; Jia, Z.-Y.; Jin, X.; Zhang, S.-B.; Li, S.-M. Influence of chitosan nanoparticles as the absorption enhancers on salvianolic acid B In vitro and In vivo evaluation. Pharmacogn. Mag. 2016, 12, 57–63.
- Smith, J.; Wood, E.; Dornish, M. Effect of Chitosan on Epithelial Cell Tight Junctions. Pharm. Res. 2004, 21, 43–49.
- Schipper, N.G.M.; Vårum, K.M.; Artursson, P. Chitosans as Absorption Enhancers for Poorly Absorbable Drugs. 1: Influence of Molecular Weight and Degree of Acetylation on Drug Transport Across Human Intestinal Epithelial (Caco-2) Cells. Pharm. Res. 1996, 13, 1686–1692.
- Thanou, M.; Verhoef, J.; Junginger, H. Oral drug absorption enhancement by chitosan and its derivatives. Adv. Drug Deliv. Rev. 2001, 52, 117–126.
- Kubbinga, M.; Augustijns, P.; García, M.A.; Heinen, C.; Wortelboer, H.M.; Verwei, M.; Langguth, P. The effect of chitosan on the bioaccessibility and intestinal permeability of acyclovir. Eur. J. Pharm. Biopharm. 2019, 136, 147–155.
- Srinivas, N.R. The Interesting Case of Acyclovir Delivered Using Chitosan in Humans: Is it a Drug Issue or Formulation Issue? Pharm. Res. 2015, 33, 543–547.
- Valdes, S.A.; Alzhrani, R.F.; Lansakara-P, D.S.P.; Cui, Z. Effect of a Solid Lipid Nanoparticle Formulation on the Bioavailability of 4-(N)-Docosahexaenoyl 2′, 2′-Difluorodeoxycytidine After Oral Administration. AAPS PharmSciTech 2020, 21, 77.
- Chen, W.; Fan, D.; Meng, L.; Miao, Y.; Yang, S.; Weng, Y.; He, H.; Tang, X. Enhancing effects of chitosan and chitosan hydrochloride on intestinal absorption of berberine in rats. Drug Dev. Ind. Pharm. 2011, 38, 104–110.
- Kontogiannidou, E.; Meikopoulos, T.; Virgiliou, C.; Bouropoulos, N.; Gika, H.; Vizirianakis, I.; Müllertz, A.; Fatouros, D.G. Towards the development of Self-Nano-Emulsifying Drug Delivery Systems (SNEDDS) containing trimethyl chitosan for the oral delivery of amphotericin B: In vitro assessment and cytocompatibility studies. J. Drug Deliv. Sci. Technol. 2020, 56, 101524.
- Qian, S.; Zhang, Q.; Wang, Y.; Lee, B.; Betageri, G.V.; Chow, M.S.; Huang, M.; Zuo, Z. Bioavailability enhancement of glucosamine hydrochloride by chitosan. Int. J. Pharm. 2013, 455, 365–373.
- Babadi, D.; Dadashzadeh, S.; Osouli, M.; Daryabari, M.S.; Haeri, A. Nanoformulation strategies for improving intestinal permeability of drugs: A more precise look at permeability assessment methods and pharmacokinetic properties changes. J. Control. Release 2020, 321, 669–709.
- Teixeira, M.; Carbone, C.; Souto, E. Beyond liposomes: Recent advances on lipid based nanostructures for poorly soluble/poorly permeable drug delivery. Prog. Lipid Res. 2017, 68, 1–11.
- Baek, J.-S.; Cho, C.-W. Surface modification of solid lipid nanoparticles for oral delivery of curcumin: Improvement of bioavailability through enhanced cellular uptake, and lymphatic uptake. Eur. J. Pharm. Biopharm. 2017, 117, 132–140.
- Porter, C.; Charman, W.N. Uptake of drugs into the intestinal lymphatics after oral administration. Adv. Drug Deliv. Rev. 1997, 25, 71–89.
- Garg, A.; Bhalala, K.; Tomar, D.S.; Wahajuddin. In-situ single pass intestinal permeability and pharmacokinetic study of developed Lumefantrine loaded solid lipid nanoparticles. Int. J. Pharm. 2017, 516, 120–130.
- Ansari, M.J.; Anwer, K.; Jamil, S.; Al-Shdefat, R.; Ali, B.E.; Ahmad, M.M. Enhanced oral bioavailability of insulin-loaded solid lipid nanoparticles: Pharmacokinetic bioavailability of insulin-loaded solid lipid nanoparticles in diabetic rats. Drug Deliv. 2015, 23, 1972–1979.
- Palmerston Mendes, L.; Pan, J.; Torchilin, V.P. Dendrimers as Nanocarriers for Nucleic Acid and Drug Delivery in Cancer Therapy. Molecules 2017, 22, 1401.
- Beezer, A.; King, A.; Martin, I.; Mitchel, J.; Twyman, L.; Wain, C. Dendrimers as potential drug carriers; encapsulation of acidic hydrophobes within water soluble PAMAM derivatives. Tetrahedron 2003, 59, 3873–3880.
- Yan, C.; Gu, J.; Lv, Y.; Shi, W.; Jing, H. Improved intestinal absorption of water-soluble drugs by acetylation of G2 PAMAM dendrimer nanocomplexes in rat. Drug Deliv. Transl. Res. 2017, 11, 408–415.
- Sadekar, S.; Thiagarajan, G.; Bartlett, K.; Hubbard, D.; Ray, A.; McGill, L.; Ghandehari, H. Poly(amido amine) dendrimers as absorption enhancers for oral delivery of camptothecin. Int. J. Pharm. 2013, 456, 175–185.
- Qi, R.; Zhang, H.; Xu, L.; Shen, W.; Chen, C.; Wang, C.; Cao, Y.; Wang, Y.; van Dongen, M.A.; He, B.; et al. G5 PAMAM dendrimer versus liposome: A comparison study on the in vitro transepithelial transport and in vivo oral absorption of simvastatin. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 1141–1151.
- Yiyun, C.; Tongwen, X. Dendrimers as Potential Drug Carriers. Part I. Solubilization of Non-Steroidal Anti-Inflammatory Drugs in the Presence of Polyamidoamine Dendrimers. Eur. J. Med. Chem. 2005, 40, 1188–1192.
- D’Emanuele, A.; Jevprasesphant, R.; Penny, J.; Attwood, D. The use of a dendrimer-propranolol prodrug to bypass efflux transporters and enhance oral bioavailability. J. Control. Release 2004, 95, 447–453.
- Choudhury, H.; Pandey, M.; Gorain, B.; Chatterjee, B.; Madheswaran, T.; Shadab; Mak, K.-K.; Tambuwala, M.; Chourasia, M.K.; Kesharwani, P. Nanoemulsions as Effective Carriers for the Treatment of Lung Cancer. In Nanotechnology-Based Targeted Drug Delivery Systems for Lung Cancer; Academic Press: Cambridge, MA, USA, 2019; pp. 217–247.
- Kawakami, K.; Yoshikawa, T.; Moroto, Y.; Kanaoka, E.; Takahashi, K.; Nishihara, Y.; Masuda, K. Microemulsion formulation for enhanced absorption of poorly soluble drugs: I. Prescription design. J. Control. Release 2002, 81, 65–74.
- Aboofazeli, R. Nanometric-Scaled Emulsions (Nanoemulsions). Iran. J. Pharm. Res. 2010, 9, 325–326.
- Isailović, T.M.; Todosijević, M.N.; Dordević, S.M.; Savić, S.D. Natural Surfactants-Based Micro/Nanoemulsion Systems for NSAIDs—Practical Formulation Approach, Physicochemical and Biopharmaceutical Characteristics/Performances. In Microsized and Nanosized Carriers for Nonsteroidal Anti-Inflammatory Drugs; Academic Press: Cambridge, MA, USA, 2017.
- Baral, K.; Song, J.-G.; Lee, S.; Bajracharya, R.; Sreenivasulu, G.; Kim, M.; Lee, K.; Han, H.-K. Enhanced Bioavailability of AC1497, a Novel Anticancer Drug Candidate, via a Self-Nanoemulsifying Drug Delivery System. Pharmaceutics 2021, 13, 1142.
- Kuncahyo, I.; Choiri, S.; Fudholi, A.; Martien, R.; Rohman, A. Development of pitavastatin-loaded super-saturable self-nano emulsion: A continues screening and optimization approach using statistical technique. J. Dispers. Sci. Technol. 2021, 1–10.
- Li, M.; Cui, J.; Ngadi, M.O.; Ma, Y. Absorption mechanism of whey-protein-delivered curcumin using Caco-2 cell monolayers. Food Chem. 2015, 180, 48–54.
- Anuar, N.; Sabri, A.H.; Effendi, T.J.B.; Hamid, K.A. Development and characterisation of ibuprofen-loaded nanoemulsion with enhanced oral bioavailability. Heliyon 2020, 6, e04570.
- Gao, F.; Zhang, Z.; Bu, H.; Huang, Y.; Gao, Z.; Shen, J.; Zhao, C.; Li, Y. Nanoemulsion improves the oral absorption of candesartan cilexetil in rats: Performance and mechanism. J. Control. Release 2011, 149, 168–174.
- Date, A.A.; Desai, N.; Dixit, R.; Nagarsenker, M. Self-nanoemulsifying drug delivery systems: Formulation insights, applications and advances. Nanomedicine 2010, 5, 1595–1616.
- Thakkar, H.P.; Khunt, A.; Dhande, R.D.; Patel, A.A. Formulation and evaluation of Itraconazole nanoemulsion for enhanced oral bioavailability. J. Microencapsul. 2015, 32, 559–569.
- Li, Y.-J.; Hu, X.-B.; Lu, X.-L.; Liao, D.-H.; Tang, T.-T.; Wu, J.-Y.; Xiang, D.-X. Nanoemulsion-based delivery system for enhanced oral bioavailability and Caco-2 cell monolayers permeability of berberine hydrochloride. Drug Deliv. 2017, 24, 1868–1873.
- Chen, S.; Zhang, J.; Wu, L.; Wu, H.; Dai, M. Paeonol nanoemulsion for enhanced oral bioavailability: Optimization and mechanism. Nanomedicine 2018, 13, 269–282.

