 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jiahong Lu | -- | 2537 | 2022-08-01 07:53:43 | | | |
| 2 | Lindsay Dong | Meta information modification | 2537 | 2022-08-01 11:32:36 | | |
Video Upload Options
Chaperone-mediated autophagy (CMA) is a protein degradation mechanism through lysosomes. By targeting the KFERQ motif of the substrate, CMA is responsible for the degradation of about 30% of cytosolic proteins, including a series of proteins associated with neurodegenerative diseases (NDs). The fact that decreased activity of CMA is observed in NDs, and ND-associated mutant proteins, including alpha-synuclein and Tau, directly impair CMA activity reveals a possible vicious cycle of CMA impairment and pathogenic protein accumulation in ND development. Given the intrinsic connection between CMA dysfunction and ND, enhancement of CMA has been regarded as a strategy to counteract ND. Indeed, genetic and pharmacological approaches to modulate CMA have been shown to promote the degradation of ND-associated proteins and alleviate ND phenotypes in multiple ND models.
1. Introduction
2. CMA as a Therapeutic Target for Neurodegenerative Diseases
2.1. Molecular Mechanism of CMA
2.1.1. CMA Substrate Recognition
2.1.2. Transportation of CMA Substrate by HSC70
2.1.3. Translocation of CMA Substrate by LAMP2A
2.2. Physiological Function of CMA
2.2.1. Starvation
2.2.2. Protein Quality Control
2.2.3. Metabolic Regulation
2.2.4. Cell Cycle Control
2.2.5. Immune Responses
2.3. Role of CMA in Neurodegenerative Diseases
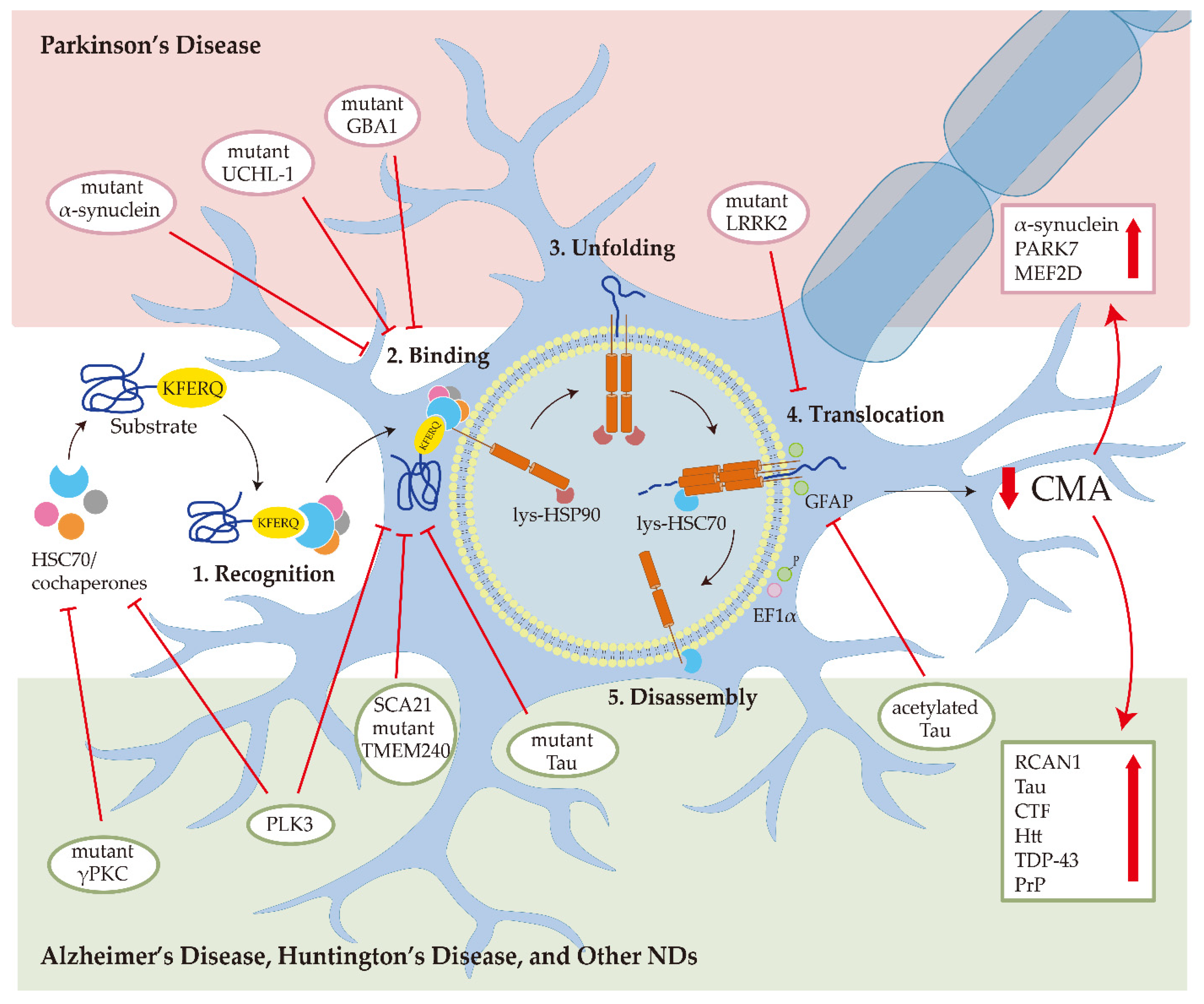
2.3.1. Parkinson’s Disease
2.3.2. Alzheimer’s Disease
2.3.3. Huntington’s Disease and Other NDs
References
- Gammon, K. Neurodegenerative disease: Brain windfall. Nature 2014, 515, 299–300.
- Lamptey, R.N.L.; Chaulagain, B.; Trivedi, R.; Gothwal, A.; Layek, B.; Singh, J. A Review of the Common Neurodegenerative Disorders: Current Therapeutic Approaches and the Potential Role of Nanotherapeutics. Int. J. Mol. Sci. 2022, 23, 1851.
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42.
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381.
- Auzmendi-Iriarte, J.; Matheu, A. Impact of Chaperone-Mediated Autophagy in Brain Aging: Neurodegenerative Diseases and Glioblastoma. Front. Aging Neurosci. 2021, 12, 630743.
- Fred Dice, J. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem. Sci. 1990, 15, 305–309.
- Kaushik, S.; Cuervo, A.M. AMPK-dependent phosphorylation of lipid droplet protein PLIN2 triggers its degradation by CMA. Autophagy 2016, 12, 432–438.
- Lv, L.; Li, D.; Zhao, D.; Lin, R.; Chu, Y.; Zhang, H.; Zha, Z.; Liu, Y.; Li, Z.; Xu, Y.; et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol. Cell 2011, 42, 719–730.
- Ferreira, J.V.; Soares, A.R.; Ramalho, J.S.; Pereira, P.; Girao, H. K63 linked ubiquitin chain formation is a signal for HIF1A degradation by Chaperone-Mediated Autophagy. Sci. Rep. 2015, 5, 10210.
- Bonam, S.R.; Ruff, M.; Muller, S. HSPA8/HSC70 in Immune Disorders: A Molecular Rheostat that Adjusts Chaperone-Mediated Autophagy Substrates. Cells 2019, 8, 849.
- Cuervo, A.M.; Dice, J.F.; Knecht, E. A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J. Biol. Chem. 1997, 272, 5606–5615.
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M. Activation of Chaperone-mediated Autophagy during Oxidative Stress. Mol. Biol. Cell 2004, 15, 4829–4840.
- Cuervo, A.M.; Dice, J.F. Unique properties of lamp2a compared to other lamp2 isoforms. J. Cell Sci. 2000, 113, 4441–4450.
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104.
- Bandyopadhyay, U.; Sridhar, S.; Kaushik, S.; Kiffin, R.; Cuervo, A.M. Identification of Regulators of Chaperone-Mediated Autophagy. Mol. Cell 2010, 39, 535–547.
- Catarino, S.; Pereira, P.; Girão, H. Molecular control of chaperone-mediated autophagy. Essays Biochem. 2017, 61, 663–674.
- Hosaka, Y.; Araya, J.; Fujita, Y.; Kuwano, K. Role of chaperone-mediated autophagy in the pathophysiology including pulmonary disorders. Inflamm. Regen. 2021, 41, 29.
- Mizushima, N.; Yamamoto, A.; Matsui, M.; Yoshimori, T.; Ohsumi, Y. In Vivo Analysis of Autophagy in Response to Nutrient Starvation Using Transgenic Mice Expressing a Fluorescent Autophagosome Marker. Mol. Biol. Cell 2004, 15, 1101–1111.
- Massey, A.C.; Kaushik, S.; Sovak, G.; Kiffin, R.; Cuervo, A.M. Consequences of the selective blockage of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2006, 103, 5805–5810.
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135.
- Cuervo, A.M.; Knecht, E.; Terlecky, S.R.; Dice, J.F. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am. J. Physiol. Cell Physiol. 1995, 269, C1200–C1208.
- Orenstein, S.J.; Cuervo, A.M. Chaperone-mediated autophagy: Molecular mechanisms and physiological relevance. Semin. Cell Dev. Biol. 2010, 21, 719–726.
- Dohi, E.; Tanaka, S.; Seki, T.; Miyagi, T.; Hide, I.; Takahashi, T.; Matsumoto, M.; Sakai, N. Hypoxic stress activates chaperone-mediated autophagy and modulates neuronal cell survival. Neurochem. Int. 2012, 60, 431–442.
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217.
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired Degradation of Mutant α-Synuclein by Chaperone-Mediated Autophagy. Science 2004, 305, 129–295.
- Issa, A.R.; Sun, J.; Petitgas, C.; Mesquita, A.; Dulac, A.; Robin, M.; Mollereau, B.; Jenny, A.; Chérif-Zahar, B.; Birman, S. The lysosomal membrane protein LAMP2A promotes autophagic flux and prevents SNCA-induced Parkinson disease-like symptoms in the Drosophila brain. Autophagy 2018, 14, 1898–1910.
- Tasset, I.; Cuervo, A.M. Role of chaperone-mediated autophagy in metabolism. FEBS J 2016, 283, 2403–2413.
- Alfaro, I.E.; Albornoz, A.; Molina, A.; Moreno, J.; Cordero, K.; Criollo, A.; Budini, M. Chaperone Mediated Autophagy in the Crosstalk of Neurodegenerative Diseases and Metabolic Disorders. Front. Endocrinol. 2018, 9, 778.
- Hallett, P.J.; Huebecker, M.; Brekk, O.R.; Moloney, E.B.; Rocha, E.M.; Priestman, D.A.; Platt, F.M.; Isacson, O. Glycosphingolipid levels and glucocerebrosidase activity are altered in normal aging of the mouse brain. Neurobiol. Aging 2018, 67, 189–200.
- Aniento, F.; Roche, E.; Cuervo, A.M.; Knecht, E. Uptake and degradation of glyceraldehyde-3-phosphate dehydrogenase by rat liver lysosomes. J. Biol. Chem. 1993, 268, 10463–10470.
- Schneider, J.L.; Suh, Y.; Cuervo, A.M. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab. 2014, 20, 417–432.
- Andrade-Tomaz, M.; de Souza, I.; Rocha, C.R.R.; Gomes, L.R. The Role of Chaperone-Mediated Autophagy in Cell Cycle Control and Its Implications in Cancer. Cells 2020, 9, 2140.
- She, H.; Mao, Z. Regulation of myocyte enhancer factor-2 transcription factors by neurotoxins. Neurotoxicology 2011, 32, 563–566.
- Yang, Q.; She, H.; Gearing, M.; Colla, E.; Lee, M.; Shacka, J.J.; Mao, Z. Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science 2009, 323, 124–127.
- Gan-Or, Z.; Dion, P.A.; Rouleau, G.A. Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy 2015, 11, 1443–1457.
- Zhang, L.; Sun, Y.; Fei, M.; Tan, C.; Wu, J.; Zheng, J.; Tang, J.; Sun, W.; Lv, Z.; Bao, J.; et al. Disruption of chaperone-mediated autophagy-dependent degradation of MEF2A by oxidative stress-induced lysosome destabilization. Autophagy 2014, 10, 1015–1035.
- Moreno-Blas, D.; Gorostieta-Salas, E.; Castro-Obregón, S. Connecting chaperone-mediated autophagy dysfunction to cellular senescence. Ageing Res. Rev. 2018, 41, 34–41.
- Zhou, D.; Li, P.; Lin, Y.; Lott, J.M.; Hislop, A.D.; Canaday, D.H.; Brutkiewicz, R.R.; Blum, J.S. Lamp-2a facilitates MHC class II presentation of cytoplasmic antigens. Immunity 2005, 22, 571–581.
- Valdor, R.; Mocholi, E.; Botbol, Y.; Guerrero-Ros, I.; Chandra, D.; Koga, H.; Gravekamp, C.; Cuervo, A.M.; Macian, F. Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat. Immunol. 2014, 15, 1046–1054.
- Zhang, J.; Huang, J.; Gu, Y.; Xue, M.; Qian, F.; Wang, B.; Yang, W.; Yu, H.; Wang, Q.; Guo, X.; et al. Inflammation-induced inhibition of chaperone-mediated autophagy maintains the immunosuppressive function of murine mesenchymal stromal cells. Cell. Mol. Immunol. 2021, 18, 1476–1488.
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340.
- Gupta, S.; Shukla, S. Non-motor symptoms in Parkinson’s disease: Opening new avenues in treatment. Curr. Res. Behav. Sci. 2021, 2, 100049.
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808.
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888.
- Li, B.; Zhang, Y.; Yuan, Y.; Chen, N. A new perspective in Parkinson’s disease, chaperone-mediated autophagy. Parkinsonism Relat. Disord. 2011, 17, 231–235.
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type α-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008, 283, 23542–23556.
- Li, G.; Yang, H.; Zhu, D.; Huang, H.; Liu, G.; Lun, P. Targeted Suppression of Chaperone-Mediated Autophagy by miR-320a Promotes α-Synuclein Aggregation. Int. J. Mol. Sci. 2014, 15, 15845–15857.
- Li, J.-Q.; Tan, L.; Yu, J.-T. The role of the LRRK2 gene in Parkinsonism. Mol. Neurodegener. 2014, 9, 47.
- Jennings, D.; Huntwork-Rodriguez, S.; Henry, A.G.; Sasaki, J.C.; Meisner, R.; Diaz, D.; Solanoy, H.; Wang, X.; Negrou, E.; Bondar, V.V.; et al. Preclinical and clinical evaluation of the LRRK2 inhibitor DNL201 for Parkinson’s disease. Sci. Transl. Med. 2022, 14, eabj2658.
- Orenstein, S.J.; Kuo, S.-H.; Tasset, I.; Arias, E.; Koga, H.; Fernandez-Carasa, I.; Cortes, E.; Honig, L.S.; Dauer, W.; Consiglio, A.; et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 2013, 16, 394–406.
- Kuo, S.-H.; Orenstein, S.; Koga, H.; Tang, G.; Kanter, E.; Alcalay, R.; Cuervo, A.M.; Sulzer, D. LRRK2 G2019S Impairs Chaperone-Mediated Autophagy in Neurons (IN2-1.001). Neurology 2013, 80, IN2-1.001-IN002-001.001.
- Kabuta, T.; Furuta, A.; Aoki, S.; Furuta, K.; Wada, K. Aberrant interaction between Parkinson disease-associated mutant UCH-L1 and the lysosomal receptor for chaperone-mediated autophagy. J. Biol. Chem. 2008, 283, 23731–23738.
- Kuo, S.-H.; Tasset, I.; Cheng, M.M.; Diaz, A.; Pan, M.-K.; Lieberman, O.J.; Hutten, S.J.; Alcalay, R.N.; Kim, S.; Ximénez-Embún, P.; et al. Mutant glucocerebrosidase impairs α-synuclein degradation by blockade of chaperone-mediated autophagy. Sci. Adv. 2022, 8, eabm6393.
- Wang, B.; Cai, Z.; Tao, K.; Zeng, W.; Lu, F.; Yang, R.; Feng, D.; Gao, G.; Yang, Q. Essential control of mitochondrial morphology and function by chaperone-mediated autophagy through degradation of PARK7. Autophagy 2016, 12, 1215–1228.
- Tang, F.-L.; Erion, J.R.; Tian, Y.; Liu, W.; Yin, D.-M.; Ye, J.; Tang, B.; Mei, L.; Xiong, W.-C. VPS35 in Dopamine Neurons Is Required for Endosome-to-Golgi Retrieval of Lamp2a, a Receptor of Chaperone-Mediated Autophagy That Is Critical for α-Synuclein Degradation and Prevention of Pathogenesis of Parkinson’s Disease. J. Neurosci. 2015, 35, 10613–10628.
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 15056.
- Block, M.L. NADPH oxidase as a therapeutic target in Alzheimer’s disease. BMC Neurosci. 2008, 9 (Suppl. S2), S8.
- Yu, W.H.; Cuervo, A.M.; Kumar, A.; Peterhoff, C.M.; Schmidt, S.D.; Lee, J.-H.; Mohan, P.S.; Mercken, M.; Farmery, M.R.; Tjernberg, L.O.; et al. Macroautophagy—A novel β-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol. 2005, 171, 87–98.
- Wang, Y.; Martinez-Vicente, M.; Krüger, U.; Kaushik, S.; Wong, E.; Mandelkow, E.-M.; Cuervo, A.M.; Mandelkow, E. Tau fragmentation, aggregation and clearance: The dual role of lysosomal processing. Hum. Mol. Genet. 2009, 18, 4153–4170.
- Bauer, P.O.; Goswami, A.; Wong, H.K.; Okuno, M.; Kurosawa, M.; Yamada, M.; Miyazaki, H.; Matsumoto, G.; Kino, Y.; Nagai, Y.; et al. Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat. Biotechnol. 2010, 28, 256–263.
- Koga, H.; Martinez-Vicente, M.; Arias, E.; Kaushik, S.; Sulzer, D.; Cuervo, A.M. Constitutive upregulation of chaperone-mediated autophagy in Huntington’s disease. J. Neurosci. 2011, 31, 18492–18505.
- Wang, H.; Tian, C.; Sun, J.; Chen, L.-N.; Lv, Y.; Yang, X.-D.; Xiao, K.; Wang, J.; Chen, C.; Shi, Q.; et al. Overexpression of PLK3 Mediates the Degradation of Abnormal Prion Proteins Dependent on Chaperone-Mediated Autophagy. Mol. Neurobiol. 2017, 54, 4401–4413.
- Soto, C.; Satani, N. The intricate mechanisms of neurodegeneration in prion diseases. Trends Mol. Med. 2011, 17, 14–24.
- Oshikawa, M.; Okada, K.; Tabata, H.; Nagata, K.I.; Ajioka, I. Dnmt1-dependent Chk1 pathway suppression is protective against neuron division. Development 2017, 144, 3303–3314.
- Seki, T.; Yoshino, K.I.; Tanaka, S.; Dohi, E.; Onji, T.; Yamamoto, K.; Hide, I.; Paulson, H.L.; Saito, N.; Sakai, N. Establishment of a novel fluorescence-based method to evaluate chaperone-mediated autophagy in a single neuron. PLoS ONE 2012, 7, e31232.



