Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Roberto Tonelli | -- | 3226 | 2022-07-29 10:43:51 | | | |
| 2 | Camila Xu | Meta information modification | 3226 | 2022-07-29 10:50:13 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Bartolucci, D.; Pession, A.; Hrelia, P.; Tonelli, R. Precision Anti-Cancer Medicines by Oligonucleotide in Clinical Reasearch. Encyclopedia. Available online: https://encyclopedia.pub/entry/25650 (accessed on 27 July 2026).
Bartolucci D, Pession A, Hrelia P, Tonelli R. Precision Anti-Cancer Medicines by Oligonucleotide in Clinical Reasearch. Encyclopedia. Available at: https://encyclopedia.pub/entry/25650. Accessed July 27, 2026.
Bartolucci, Damiano, Andrea Pession, Patrizia Hrelia, Roberto Tonelli. "Precision Anti-Cancer Medicines by Oligonucleotide in Clinical Reasearch" Encyclopedia, https://encyclopedia.pub/entry/25650 (accessed July 27, 2026).
Bartolucci, D., Pession, A., Hrelia, P., & Tonelli, R. (2022, July 29). Precision Anti-Cancer Medicines by Oligonucleotide in Clinical Reasearch. In Encyclopedia. https://encyclopedia.pub/entry/25650
Bartolucci, Damiano, et al. "Precision Anti-Cancer Medicines by Oligonucleotide in Clinical Reasearch." Encyclopedia. Web. 29 July, 2022.
Copy Citation
Oligonucleotide therapeutics enable a direct targeting of the gene by acting at the level of the RNA or at the level of the DNA, based on the Watson-Crick complementary rule of binding.
cancer therapy
new anti-cancer drugs
precision medicine
undruggable targets
1. Oligonucleotide Therapeutics as New Targeted Anti-Cancer Drugs for Challenging or Undruggable Proteins
The molecular targets for therapy could be divided in two major categories, namely, druggable and undruggable. “Druggability” implies that the target molecule must have structures that should allocate the specific binding and inhibition by low-molecular-weight compounds. Typically, a protein is considered druggable if it contains a cavity, usually a well-defined catalytic cleft. It is estimated that almost 70% of the human proteins [1] are considered difficult to be targeted, including the transcription factors that are widely thought to be undruggable due to the lack of catalytic clefts and the much-sought drug-binding pockets. To date, targeting the relevant transcription factors with small molecular compounds remains challenging. In particular, relevant examples are the members of the MYC family and RAS family of oncogenes, that account for amongst the highest rate of mutations in cancers and define aggressive tumor behaviors [2][3]. The strategies that propose the indirect blocking of transcription factors by targeting their upstream or downstream pathway genes could result in less efficacy and also specific side effects in healthy cells. New strategies are urgently needed to generate drugs able to tackle the undruggable targets in cancer. In this respect, oligonucleotide therapeutics enable a direct targeting of the gene by acting at the level of the RNA or at the level of the DNA, based on the Watson-Crick complementary rule of binding. Different classes of oligonucleotides have been developed since the first use in clinical was proposed in the 1970s, but, in particular, antisense oligonucleotides (ASOs), small interference RNAs (siRNAs), and microRNAs (miRNAs) imposed their presence as the most representative for clinical development and therapeutic application [4][5]. For a better understanding of the impact and the relevance of each group, researchers performed a research study in clinicaltrials.gov (accessed on 1 May 2022) using “Cancer” as the keyword and focusing the attention on clinical trials available for each class of oligonucleotide in analysis.
As expected, the ASOs are the most represented oligonucleotides, accounting for 66% of all of the compounds analyzed (75% of clinical trials), and are the only class with some compounds in clinical phase II or III (Figure 1 and Table 1). Interestingly, only 12% of the clinical studies are recruiting or active, and the majority are completed (59%), or in other status (29%). From a structural point of view, the ASOs range in size from 12 to 30 nucleotides, are single stranded, and work through the classic Watson-Crick base pairing and can act as both gene expression inhibitors or splicing modulators [6][7].
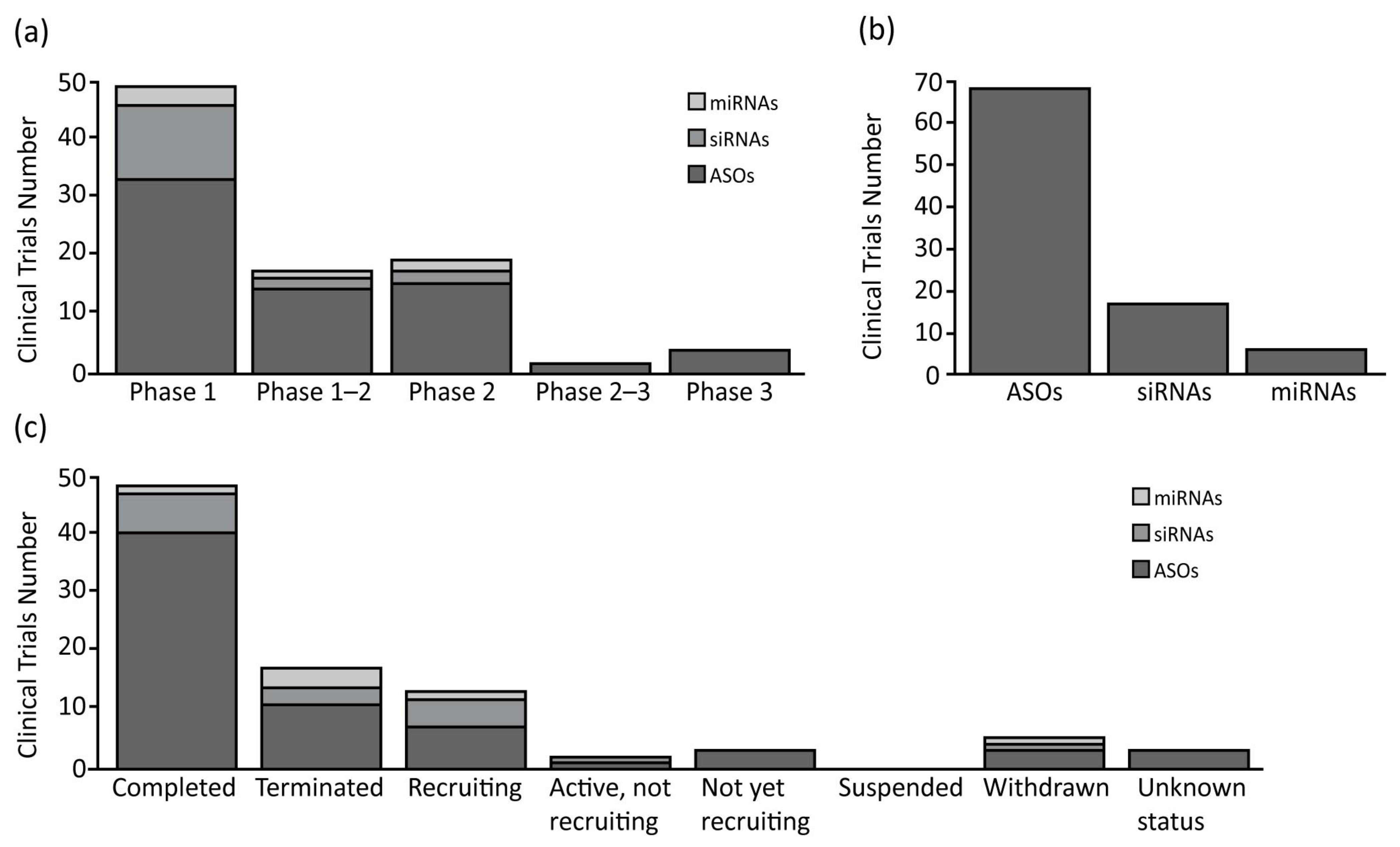
Figure 1. Summary of clinical trials therapies using oligonucleotide in oncology. (a) Antisense oligonucleotides (ASOs), small interfering RNAs (siRNAs) and microRNAs (miRNAs) number of clinical trials divided for clinical phases; (b) Total clinical trials number for each class of oligonucleotide analyzed; (c) ASOs, siRNAs and miRNAs number of clinical trials divided for clinical status condition.
Table 1. Summary of oligonucleotides in clinical trials for oncology.
| Oligonucleotide | Target | Drug Type | Cancer Type | Clinical Phase | Clinical Trials ID |
|---|---|---|---|---|---|
| WGI-0301 | AKT1 | ASO | Advanced Solid Tumors | Phase 1 | NCT05267899 |
| AZD5312 | AR | ASO | Advanced Solid Tumors with Androgen Receptor Pathway as a Potential Factor | Phase 1 | NCT02144051 |
| BP1002 (L-Bcl-2 antisense oligonucleotide) | BCL-2 | ASO | Mantle Cell Lymphoma|Peripheral T-cell Lymphoma (PTCL)|Cutaneous T-cell Lymphoma (CTCL)|Chronic Lymphocytic Leukemia (CLL)|Small Lymphocytic Lymphoma (SLL)|Follicular Lymphoma|Marginal Zone Lymphoma|Hodgkin Lymphoma|Waldenstrom Macroglobulinemia|DLBCL | Phase 1 | NCT04072458 |
| PNT2258 | BCL-2 | ASO | Cancer|Lymphoma|Prostate Cancer|Melanoma | Phase 1 | NCT01191775 |
| LErafAON | CRAF | ASO | Neoplasms | Phase 1 | NCT00024648|NCT00024661|NCT00100672 |
| AZD8701 | FOXP3 | ASO | Clear Cell Renal Cell Cancer|Non-Small-Cell Lung Cancer|Triple Negative Breast Neoplasms|Squamous Cell Cancer of Head and Neck|Small-Cell Lung Cancer|Gastroesophageal Cancer|Melanoma|Cervical Cancer|Advanced Solid Tumors | Phase 1 | NCT04504669 |
| BP1001-A (Liposomal Grb2 Antisense Oligonucleotide) | GRB-2 | ASO | Solid Tumor, Adult|Carcinoma, Ovarian Epithelial|Fallopian Tube Neoplasms|Endometrial Cancer|Peritoneal Cancer|Solid Tumor | Phase 1 | NCT04196257 |
| EZN-2968 | HIF-1α | ASO | Neoplasms|Liver Metastases|Carcinoma|Lymphoma | Phase 1 | NCT01120288|NCT00466583 |
| AZD4785 | KRAS | ASO | Non-Small-Cell Lung Cancer|Advanced Solid Tumors | Phase 1 | NCT03101839 |
| TASO-001 | TGF-β2 | ASO | Solid Tumor | Phase 1 | NCT04862767 |
| SD-101 (CpG Oligonucleotide) | TLR9 | ASO | Advanced Malignant Solid Neoplasm|Extracranial Solid Neoplasm|Metastatic Malignant Solid Neoplasm | Phase 1 | NCT03831295 |
| CpG7909 (PF3512676) | TLR9 | ASO | Intraocular Melanoma|Malignant Conjunctival Neoplasm|Melanoma (Skin) | Phase 1 | NCT00471471 |
| ISS 1018 (CpG ODN) | TLR9 | ASO | Colorectal Neoplasms | Phase 1 | NCT00403052 |
| IONIS-STAT3Rx (AZD9150) | STAT3 | ASO | Hepatocellular Carcinoma|Ovarian Cancer|Ascites|Gastrointestinal Cancer|Advanced Cancers|DLBCL|Lymphoma | Phase 1|2 | NCT01839604|NCT02417753|NCT01563302|NCT02549651 |
| ISIS 183750 | eIF4E | ASO | Colorectal Neoplasms|Colorectal Carcinoma|Colorectal Tumors | Phase 1|Phase 2 | NCT01675128 |
| Apatorsen (OGX-427) | HSP-27 | ASO | Squamous Cell Lung Cancer|Bladder Cancer|Urothelial Carcinoma|Prostate Cancer | Phase 1|Phase 2 | NCT02423590|NCT00959868|NCT00487786|NCT01780545|NCT01120470 |
| GTI-2040 | R2 subunit of RNR | ASO | Carcinoma, Renal Cell|Metastases, Neoplasm | Phase 1|Phase 2 | NCT00056173 |
| Aezea (Cenersen) | TP53 | ASO | Myelodysplastic Syndromes|Acute Myelogenous Leukemia | Phase 1|Phase 2 | NCT02243124|NCT00967512 |
| VEGF-Antisense Oligonucleotide | VEGF | ASO | Mesothelioma | Phase 1|Phase 2 | NCT00668499 |
| AEG35156 | XIAP | ASO | Human Mammary Carcinoma|Carcinoma|Pancreas|Non-Small-Cell Lung | Phase 1|Phase 2 | NCT00385775|NCT00558545|NCT00557596|NCT00558922 |
| XIAP Antisense | XIAP | ASO | Leukemia, Myelomonocytic, Acute | Phase 1|Phase 2 | NCT00363974 |
| Oblimersen (G3139) | BCL-2 | ASO | Lymphoma|Prostate Cancer|Lung Cancer|Melanoma (Skin)|Colorectal Cancer|Breast Cancer | Phase 1|Phase 2|Phase 3 | NCT00070083|NCT00080847|NCT00017251|NCT00070343|NCT00016263|NCT00017602|NCT00085228|NCT00030641|NCT00063934|NCT00004870|NCT00005032|NCT00054548|NCT00543231|NCT00543205|NCT00636545|NCT00078234|NCT00021749|NCT00024440|NCT00059813 |
| G4460 (c-myb antisense oligonucleotide) | C-MYB | ASO | Hematologic Malignancies | Phase 2 | NCT00780052|NCT00002592 |
| ISIS 5132 | CRAF | ASO | Breast Cancer | Phase 2 | NCT00003236 |
| BP1001 (Liposomal Grb2 Antisense Oligonucleotide) | GRB-2 | ASO | Recurrent Adult Acute Myeloid Leukemia|Acute Lymphoblastic Leukemia|Myelodysplastic Syndrome|Ph1 Positive CML | Phase 2 | NCT02923986|NCT02781883|NCT01159028 |
| IGV-001 Cell Immunotherapy | IGF type 1 receptor | ASO | Glioblastoma Multiforme|Glioblastoma | Phase 2 | NCT04485949 |
| ISIS 3521 | PKCα | ASO | Breast Cancer | Phase 2 | NCT00003236 |
| STP705 | TGF-β1 and COX-2 | ASO | Squamous Cell Carcinoma in Situ | Phase 2 | NCT04844983 |
| CpG-ODN | TLR9 | ASO | Glioblastoma|Lung Cancer|Hepatocellular Carcinoma|Solid Tumor | Phase 2 | NCT00190424|NCT04952272 |
| Custirsen (OGX-011) | clusterin | ASO | Prostate Cancer|Bladder Cancer|Breast Cancer|Kidney Cancer|Lung Cancer|Ovarian Cancer|Unspecified Adult Solid Tumor | Phase 2|Phase 3 | NCT00054106|NCT00258375|NCT00471432|NCT01083615 |
| INT-1B3 | JNK1 | miRNA | Solid Tumor | Phase 1 | NCT04675996 |
| TargomiRs | Multiple oncogenes, including BCL2, MCL1, CCND1, and WNT3A | miRNA | Malignant Pleural Mesothelioma|Non-Small-Cell Lung Cancer | Phase 1 | NCT02369198 |
| MRX34 | 30 unique oncogenes, including but not limited to MET, MYC, PDGFR-a, CDK4/6 and BCL2 | miRNA | Melanoma | Phase 1|Phase 2 | NCT01829971|NCT02862145 |
| Cobomarsen (MRG-106) | mir-155 | miRNA | Cutaneous T-Cell Lymphoma/Mycosis Fungoides | Phase 2 | NCT03837457|NCT03713320 |
| siRNA-transfected peripheral blood mononuclear cells APN401 | CBLB | siRNA | Metastatic Malignant Neoplasm in the Brain|Metastatic Solid Neoplasm|Recurrent Colorectal Carcinoma|Recurrent Melanoma|Recurrent Pancreatic Cancer|Recurrent Renal Cell Cancer | Phase 1 | NCT03087591|NCT02166255 |
| EphA2-targeting DOPC-encapsulated siRNA | EPHA2 | siRNA | Advanced Malignant Solid Neoplasm | Phase 1 | NCT01591356 |
| NBF-006 | GSTP | siRNA | Non-Small-Cell Lung Cancer|Pancreatic Cancer|Colorectal Cancer | Phase 1 | NCT03819387 |
| Mesenchymal Stromal Cells-derived Exosomes with KRAS G12D siRNA | KRASG12D | siRNA | Metastatic Pancreatic Adenocarcinoma|Pancreatic Ductal Adenocarcinoma | Phase 1 | NCT03608631 |
| Proteasome siRNA and tumor antigen RNA-transfected dendritic cells | LMP2, LMP7, MECL1 | siRNA | Metastatic Melanoma|Absence of CNS Metastases | Phase 1 | NCT00672542 |
| CALAA-01 | M2 subunit of ribonucleotide reductase (R2) | siRNA | Cancer|Solid Tumor | Phase 1 | NCT00689065 |
| TKM-080301 | PLK1 | siRNA | Colorectal Cancer with Hepatic Metastases|Pancreas Cancer with Hepatic Metastase|Gastric Cancer With Hepatic Metastases|Breast Cancer With Hepatic | Phase 1 | NCT01437007 |
| SLN124 | TMPRSS6 | siRNA | Non-transfusion-dependent Thalassemia|Low Risk Myelodysplastic Syndrome | Phase 1 | NCT04176653 |
| DCR-MYC | MYC | siRNA | Solid Tumors|Multiple Myeloma|Non-Hodgkins Lymphoma|Pancreatic Neuroendocrine Tumors|PNET|NHL| Hepatocellular Carcinoma |
Phase 1|Phase 2 | NCT02110563|NCT02314052 |
| Atu027 | PNK3 | siRNA | Advanced Solid Tumors|Carcinoma, Pancreatic Ductal | Phase 1|Phase 2 | NCT00938574|NCT01808638 |
| siG12D LODER | KRASG12D | siRNA | Pancreatic Ductal Adenocarcinoma|Pancreatic Cancer | Phase 2 | NCT01188785|NCT01676259 |
| STP705 | TGF-β1, COX-2 mRNA | siRNA | Squamous Cell Carcinoma in Situ | Phase 2 | NCT04844983 |
| CpG-STAT3 siRNA CAS3/SS3 | TLR9 and STAT3 | siRNA | B-Cell Non-Hodgkin Lymphoma|Diffuse Large B-Cell Lymphoma|Follicular Lymphoma|Mantle Cell Lymphoma|Marginal Zone Lymphoma|Small Lymphocytic Lymphoma | Phase 1 | NCT04995536 |
The second most common oligonucleotide class is represented by the siRNAs, accounting for 25% of the compounds. The analysis shows how they are the subject of almost the same number of clinical trials as the miRNAs, but with a higher number of trials in phase I (76% of total siRNAs’ clinical trials). In relation to the clinical trial status, the siRNAs show 35% active/recruiting trials, and 18% of the trials terminated with no suspension (Figure 1 and Table 1). The siRNAs are the longest oligonucleotides in size (20–25 nt), they are double stranded, and work in complex with RISC to post-transcriptionally silence the target gene expressions [8]. These compounds can be chemically synthesized, maintaining the characteristics needed for the proper activation of the enzymatic mRNA degradation, allowing their use as therapeutic compounds [9].
Finally, the miRNAs represent the smallest class with only four compounds in clinical trial (9%). Despite their number, these compounds are well represented in clinical trials, as mentioned before, and are equally distributed among phases 1 and 2. Furthermore, a relevant number of clinical trials are recruiting (17%), while others are now terminated (50%), or withdrawn (17%) (Figure 1 and Table 1). The miRNAs are small (18–25 nt) single stranded non-coding RNAs, containing usually sequences complementary to one or more of the target RNAs [10]. They work similarly to the siRNAs, activating the RISC complex after formation of miRNAs duplex [11]. The miRNAs can also be synthesized by mimicking their normal biological function, to be redirected against specific targets for therapeutic purposes [12].
2. Oligonucleotide Therapeutics Targeting the MYC Gene Family
The MYC family is composed of c-MYC, MYCN, and MYCL [13] The MYC gene family encodes for the basic helix-loop-helix-leucine zipper (bHLH-LZ) transcription factor proteins which exhibit a high-structural homology, including highly conserved Myc boxes (MB) and a basic region (BR), helix-loop-helix (HLH) and leucin zipper (LZ) motifs [14][15]. The MYC gene family is highly involved in tumors in which they are often dysregulated and mutated (manly by translocations or gene amplification), resulting in overexpression and association with tumor aggressiveness and poor prognosis [16]. The proteins that originate from the MYC gene family are mainly considered undruggable with the conventional approaches that rely on small molecules.
2.1. Oligonucleotide Therapeutics Targeting MYC
MYC has been proposed as an important oncogenic target for its role in cell proliferation and survival, angiogenesis, metastasis, drug resistance, and poor patient prognosis [17][18]. MYC-targeted oligonucleotide therapeutics, based on a small interfering RNA lipid-based nanoparticle (DCR-MYC, Dicerna Pharmaceuticals), to inhibit the oncogene MYC at the level of the mRNA, was developed to treat various cancer types, including hepatocellular carcinoma (HCC), solid tumors, lymphoma, or multiple myeloma. The liposomal delivery system of the DCR-MYC is based on EnCore Dicerna’s proprietary technology, due to its specific Envelope and Core lipid contents. For targeting MYC, Dicer-substrate small interfering RNA (DsiRNA) was used in the drug formulation. DsiRNAs, longer duplex RNAs, are Dicer substrates to be subsequently processed into small interfering RNAs (siRNAs), and have an increased potency in RNA interfering processing [19]. The data from the phase I—dose-escalation study indicated that DCR-MYC presents good clinical and metabolic responses in patients at a variety of dose levels [20]. However, phase I (clinicaltrials.gov (accessed on 1 May 2022) NCT02110563) and phase Ib/2 trials (clinicaltrials.gov (accessed on 1 May 2022) NCT02314052) were terminated on the sponsor’s decision, due to a lack of the gene-silencing effectiveness that was anticipated by the company [21].
2.2. Oligonucleotide Therapeutics Targeting MYCN
The MYCN oncogene is a well-known driver of different, highly aggressive tumors (including Neuroblastoma, Small-Cell Lung Cancer, Rhabdomyosarcoma), where it is dysregulated and amplified and is strongly associated with poor survival prognosis [22][23]. MYCN overexpression reprograms the tumor cells towards a stem-like phenotype that promotes proliferation and cell growth, while inhibiting cell differentiation and apoptosis. It also favors immune escape, invasion, metastases, and angiogenesis [24][25]. Interestingly, MYCN is expressed during embryogenesis and has a highly restricted pattern of expression in normal cells after birth [26]. All of these factors make the N-Myc protein a promising target for a tumor-specific therapy. However, inhibitors against the N-Myc protein have, to-date, largely failed and have led to N-Myc being currently considered to be an undruggable target [27].
It has been demonstrated that an alternative approach concerns specific gene expression inhibition at the level of DNA through a MYCN-specific antigene peptide nucleic acid (agPNA) oligonucleotide [28][29]. The antigene oligonucleotide approach (via persistent blocking at the level of transcription) has shown advantages in blocking translation by the antisense oligonucleotide strategies. The peptide nucleic acids (PNAs) have shown promising results as antigenes, due to their resistance to proteases and nucleases and their ability to potently and specifically bind the target DNA [30][31]. Differing from the use of antisense oligonucleotides, which inhibit mRNA translation, the antigene approach involves binding to the chromosomal DNA, resulting in the inhibition of transcription. By persistently blocking the transcription, the antigene oligonucleotides showed higher efficacy compared with antisense oligonucleotides [28][30]. Antigene therapy by targeting MYCN transcription has great potential in treating MYCN-expressing tumors, as was previously demonstrated in the preclinical treatment of neuroblastoma and rhabdomyosarcoma by MYCN-specific agPNA [29][30]. Neuroblastoma (NB) is the deadliest pediatric tumor. Approximately 25% of patients with a NB diagnosis present with MYCN amplification (MNA), which is linked to a poor prognosis, metastasis, and recurrence [32][33]. It has been shown that BGA002, a new and highly improved agPNA oligonucleotide, is able to specifically target a unique sequence on the human MYCN gene [34]. BGA002 showed a specific, dose-dependent decrease in the MYCN mRNA and protein, while decreasing the viability in a panel of 20 NB cell lines, followed by the block of different MYCN tumorigenic alterations, and to the anti-tumor efficacy of BGA002 in vivo in a MNA NB mouse model [34]. Moreover, while MYCN drives a tumor immunosuppressive environment, which impacts survival in several MYCN-positive tumors, the block of MYCN by the anti-MYCN BGA002 is able to reactivate and restore the effectiveness of the natural killer immune cells against NB [35]. It has been also found that BGA002 restores the retinoic acid (RA) response, leading to a differentiation or apoptosis in the MNA NB and also to a significant increase in survival in a mouse model of MNA-NB [36].
3. Oligonucleotide Therapeutics Targeting the RAS Gene Family
Ras proteins regulate the activation of different patterns strongly involved in cancer, such as cell proliferation, differentiation, and survival [37]. This family of proteins is encoded by three ubiquitously expressed genes, HRAS, KRAS, and NRAS, that share most of their sequence and function [38]. In normal conditions, they act as the activator of more than 20 different proteins from different effectors’ families [39], so their constitutive activation may cause a deregulation of many cell functions and lead to cancer. In particular, the frequency and the pattern of the gene mutations can associate different Ras genes with different cancer types [40]. While each Ras member is involved in cancer, KRAS is surely the major cancer-causing isoform, accounting for 75% of all Ras-associated tumors [41]. The other isoforms NRAS (17%) and HRAS (7%) account for only a small subset of cancer types [41]. In the past years, many attempts were performed to develop direct Ras gene family inhibitors, but the protein structure showed characteristics that were not very compatible with the small molecules’ approach [42]. As the same post-translational approach was ineffective, due to isoform-specific differences [43], so a more specific approach was needed in consideration of the relevance of the KRAS specific isoform to the others. In particular, the oligonucleotides have a promising therapeutic potential as mutant-specific RAS inhibitors, active against any major mutation.
Oligonucleotide Therapeutics Targeting KRAS
The KRAS-targeted siRNA-polymeric nanoparticles for local therapy, siG12D-LODER, were designed by Silenseed Ltd. for patients with locally advanced pancreatic cancer. The biodegradable polymer matrix, Local Drug EluteR (LODER), was used to release the G12D-mutated KRAS-targeted siRNA locally within a pancreatic tumor microenvironment for controlled and prolonged delivery [44]. The LODER matrix consists of a copolymer of poly lactic-co-glycolic acid (PLGA) of a high molecular weight greater than 50 kD. The siG12D-LODER was designed to be properly inserted and placed into the tumor using a standard biopsy procedure [45][46]. A slow and stable release of siRNA from siG12D-LODER over a few months was demonstrated when incubated in PBS. The siG12D-LODER remarkably suppressed the growth of pancreatic tumors in both subcutaneous and orthotopic, xenograft, and syngraft mouse models, without causing any toxicity [44]. In another preclinical study, following subcutaneous implantation of the siG12D-LODER, all of the rats exhibited local and systemic safety and tolerability, without any adverse effects or deaths [46]. A phase I clinical trial was conducted by injection of the siG12D-LODER drug into patients via the endoscopic ultrasound (EUS) biopsy needle (clinicaltrials.gov (accessed on 1 May 2022) NCT01188785).
The ASO strategy has also been proposed for the treatment of the KRAS mutation in cancer. AZD4785 is a cEt-modified ASO [47], complementary to a KRAS mRNA sequence, developed by Ionis in collaboration with Astra Zeneca. Interestingly, the advanced chemistry of this compound and the resulting potency, allowed its use without any delivery agent in the first preclinical studies. As expected, AZD4785 is able to directly downregulate KRAS mRNA at the nM level in vitro (IC50 10 nM), and is also able to increase survival and reduce tumor growth in a mouse model of lung cancer [48].
4. Oligonucleotide Therapeutics Targeting STAT3
STAT3 is a protein activated by members of the JAK family through phosphorylation [49]. In its phosphorylated form, STAT3 dimerizes and works in the nucleus as a transcription factor involved in cell proliferation, development, differentiation, inflammation, and apoptosis. Constitutive activation can be found in several types of human cancer [50][51], and it is able to increase the level of different cancer-related molecules, such as surviving, Bcl-XL, cyclin D1/D2, C-Myc, Mcl-1, and vascular endothelial growth factor (VEGF), favoring tumorigenic progression [52][53]. For this reason, STAT3 represents one of the most interesting targets for therapeutics in oncology, and in the past years many strategies were developed to effectively inhibit STAT3 expression. For example, synthetic inhibitors, such as CDDO-Me or FLLL32, can reduce the activity of the protein downstream, blocking the JAK/STAT3 interaction or inhibiting the DNA binding process but cannot overcome the issue related to the overall aspect of STAT3 overexpression [54][55]. In a different way, oligonucleotide compounds are able to specifically target STAT3 reducing the protein expression, directly downregulating the mRNA. A new compound, such as AZD9150 (ISIS 481464), a 16-nucleotide next generation chemistry antisense oligonucleotide [56], or CpG-Stat3 siRNA, a conjugate of an oligonucleotide TLR9 agonist linked to a STAT3 siRNA [57], are, in fact, designed to specifically reach this aim. In particular, the combined use of synthetic oligonucleotide agonists for TLRs and siRNA target-specific, is a novel and potent strategy that can achieve both target delivery enhancement of siRNA to immune cells and antitumor immune response activation [57].
5. Oligonucleotide Therapeutics Targeting BCL-2
BCL-2 is one of the first-discovered regulators of the apoptosis process. This gene is overexpressed by the translocation t(14;18) in B-cell lymphoma and is implicated in many different cancers, such as melanoma, breast, and lung carcinoma [58]. BCL-2 is not only involved in the neoplastic development, but also in the resistance mechanism to cancer treatment [59]. In this context, the therapies pointed at BCL-2 inhibition can be considered crucial to overcome resistance to the common strategies for cancer treatments [60]. Oblimersen (G3139) is certainly the most studied BCL-2 inhibitor and was involved in many clinical studies (Table 1). Unfortunately, despite its high efficacy in vitro and in vivo preclinical studies [61][62], the clinical studies showed how it is important to administer this compound in combination to achieve significant results. Phase 3 clinical studies (data not shown) are indeed available, but only for this form of application. It is important to underline that other BCL-2 inhibitors have been developed, such as BP1002 [63] and PNT2258 [64]. Both of them are still in clinical phase I trials; the first active, and the second successfully completed, and they represent promising compounds for the future of the Bcl-2 inhibitor.
References
- Wang, C.; Fang, H.; Zhang, J.; Gu, Y. Targeting “Undruggable” c-Myc Protein by Synthetic Lethality. Front. Med. 2021, 15, 541–550.
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS Mutation: From Undruggable to Druggable in Cancer. Signal Transduct. Target. Ther. 2021, 6, 386.
- Duffy, M.J.; O’Grady, S.; Tang, M.; Crown, J. MYC as a Target for Cancer Treatment. Cancer Treat. Rev. 2021, 94, 102154.
- Moumné, L.; Marie, A.-C.; Crouvezier, N. Oligonucleotide Therapeutics: From Discovery and Development to Patentability. Pharmaceutics 2022, 14, 260.
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous Sarcoma Viral RNA Translation by a Specific Oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 285–288.
- Quemener, A.M.; Bachelot, L.; Forestier, A.; Donnou-Fournet, E.; Gilot, D.; Galibert, M. The Powerful World of Antisense Oligonucleotides: From Bench to Bedside. WIREs RNA 2020, 11, e1594.
- Deleavey, G.F.; Damha, M.J. Designing Chemically Modified Oligonucleotides for Targeted Gene Silencing. Chem. Biol. 2012, 19, 937–954.
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a Bidentate Ribonuclease in the Initiation Step of RNA Interference. Nature 2001, 409, 363–366.
- Thompson, J.D. Clinical Development of Synthetic SiRNA Therapeutics. Drug Discov. Today Ther. Strateg. 2013, 10, e133–e138.
- MacFarlane, L.-A.; Murphy, P.R. MicroRNA: Biogenesis, Function and Role in Cancer. Curr. Genom. 2010, 11, 537–561.
- Ha, M.; Kim, V.N. Regulation of MicroRNA Biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524.
- Matsuyama, H.; Suzuki, H.I. Systems and Synthetic MicroRNA Biology: From Biogenesis to Disease Pathogenesis. Int. J. Mol. Sci. 2019, 21, 132.
- Ryan, K.M.; Birnie, G.D. Myc Oncogenes: The Enigmatic Family. Biochem. J. 1996, 314, 713–721.
- Sammak, S.; Hamdani, N.; Gorrec, F.; Allen, M.D.; Freund, S.M.V.; Bycroft, M.; Zinzalla, G. Crystal Structures and Nuclear Magnetic Resonance Studies of the Apo Form of the C-MYC:MAX BHLHZip Complex Reveal a Helical Basic Region in the Absence of DNA. Biochemistry 2019, 58, 3144–3154.
- Beaulieu, M.-E.; Castillo, F.; Soucek, L. Structural and Biophysical Insights into the Function of the Intrinsically Disordered Myc Oncoprotein. Cells 2020, 9, 1038.
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35.
- Knies-Bamforth, U.E.; Fox, S.B.; Poulsom, R.; Evan, G.I.; Harris, A.L. C-Myc Interacts with Hypoxia to Induce Angiogenesis In Vivo by a Vascular Endothelial Growth Factor-Dependent Mechanism. Cancer Res. 2004, 64, 6563–6570.
- Ma, L.; Young, J.; Prabhala, H.; Pan, E.; Mestdagh, P.; Muth, D.; Teruya-Feldstein, J.; Reinhardt, F.; Onder, T.T.; Valastyan, S.; et al. MiR-9, a MYC/MYCN-Activated MicroRNA, Regulates E-Cadherin and Cancer Metastasis. Nat. Cell Biol. 2010, 12, 247–256.
- Doré-Savard, L.; Roussy, G.; Dansereau, M.-A.; Collingwood, M.A.; Lennox, K.A.; Rose, S.D.; Beaudet, N.; Behlke, M.A.; Sarret, P. Central Delivery of Dicer-Substrate SiRNA: A Direct Application for Pain Research. Mol. Ther. 2008, 16, 1331–1339.
- Tolcher, A.W.; Papadopoulos, K.P.; Patnaik, A.; Rasco, D.W.; Martinez, D.; Wood, D.L.; Fielman, B.; Sharma, M.; Janisch, L.A.; Brown, B.D.; et al. Safety and Activity of DCR-MYC, a First-in-Class Dicer-Substrate Small Interfering RNA (DsiRNA) Targeting MYC, in a Phase I Study in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2015, 33, 11006.
- Whitfield, J.R.; Beaulieu, M.-E.; Soucek, L. Strategies to Inhibit Myc and Their Clinical Applicability. Front. Cell Dev. Biol. 2017, 5, 10.
- Brodeur, G.M.; Seeger, R.C.; Schwab, M.; Varmus, H.E.; Bishop, J.M. Amplification of N-Myc in Untreated Human Neuroblastomas Correlates with Advanced Disease Stage. Science 1984, 224, 1121–1124.
- Seeger, R.C.; Brodeur, G.M.; Sather, H.; Dalton, A.; Siegel, S.E.; Wong, K.Y.; Hammond, D. Association of Multiple Copies of the N-Myc Oncogene with Rapid Progression of Neuroblastomas. N. Engl. J. Med. 1985, 313, 1111–1116.
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415.
- Dang, C.V.; Kim, J.; Gao, P.; Yustein, J. The Interplay between MYC and HIF in Cancer. Nat. Rev. Cancer 2008, 8, 51–56.
- Zimmerman, K.A.; Yancopoulos, G.D.; Collum, R.G.; Smith, R.K.; Kohl, N.E.; Denis, K.A.; Nau, M.M.; Witte, O.N.; Toran-Allerand, D.; Gee, C.E.; et al. Differential Expression of Myc Family Genes during Murine Development. Nature 1986, 319, 780–783.
- Fletcher, J.I.; Ziegler, D.S.; Trahair, T.N.; Marshall, G.M.; Haber, M.; Norris, M.D. Too Many Targets, Not Enough Patients: Rethinking Neuroblastoma Clinical Trials. Nat. Rev. Cancer 2018, 18, 389–400.
- Janowski, B.A.; Kaihatsu, K.; Huffman, K.E.; Schwartz, J.C.; Ram, R.; Hardy, D.; Mendelson, C.R.; Corey, D.R. Inhibiting Transcription of Chromosomal DNA with Antigene Peptide Nucleic Acids. Nat. Chem. Biol. 2005, 1, 210–215.
- Tonelli, R.; Purgato, S.; Camerin, C.; Fronza, R.; Bologna, F.; Alboresi, S.; Franzoni, M.; Corradini, R.; Sforza, S.; Faccini, A.; et al. Anti-Gene Peptide Nucleic Acid Specifically Inhibits MYCN Expression in Human Neuroblastoma Cells Leading to Cell Growth Inhibition and Apoptosis. Mol. Cancer Ther. 2005, 4, 779–786.
- Tonelli, R.; McIntyre, A.; Camerin, C.; Walters, Z.S.; Di Leo, K.; Selfe, J.; Purgato, S.; Missiaglia, E.; Tortori, A.; Renshaw, J.; et al. Antitumor Activity of Sustained N-Myc Reduction in Rhabdomyosarcomas and Transcriptional Block by Antigene Therapy. Clin. Cancer Res. 2012, 18, 796–807.
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-Selective Recognition of DNA by Strand Displacement with a Thymine-Substituted Polyamide. Science 1991, 254, 1497–1500.
- Campbell, K.; Gastier-Foster, J.M.; Mann, M.; Naranjo, A.H.; Van Ryn, C.; Bagatell, R.; Matthay, K.K.; London, W.B.; Irwin, M.S.; Shimada, H.; et al. Association of MYCN Copy Number with Clinical Features, Tumor Biology, and Outcomes in Neuroblastoma: A Report from the Children’s Oncology Group: MYCN Copy Number in Neuroblastoma. Cancer 2017, 123, 4224–4235.
- Qing, G.; Li, B.; Vu, A.; Skuli, N.; Walton, Z.E.; Liu, X.; Mayes, P.A.; Wise, D.R.; Thompson, C.B.; Maris, J.M.; et al. ATF4 Regulates MYC-Mediated Neuroblastoma Cell Death upon Glutamine Deprivation. Cancer Cell 2012, 22, 631–644.
- Montemurro, L.; Raieli, S.; Angelucci, S.; Bartolucci, D.; Amadesi, C.; Lampis, S.; Scardovi, A.L.; Venturelli, L.; Nieddu, G.; Cerisoli, L.; et al. A Novel MYCN-Specific Antigene Oligonucleotide Deregulates Mitochondria and Inhibits Tumor Growth in MYCN-Amplified Neuroblastoma. Cancer Res. 2019, 79, 6166–6177.
- Raieli, S.; Di Renzo, D.; Lampis, S.; Amadesi, C.; Montemurro, L.; Pession, A.; Hrelia, P.; Fischer, M.; Tonelli, R. MYCN Drives a Tumor Immunosuppressive Environment Which Impacts Survival in Neuroblastoma. Front. Oncol. 2021, 11, 625207.
- Lampis, S.; Raieli, S.; Montemurro, L.; Bartolucci, D.; Amadesi, C.; Bortolotti, S.; Angelucci, S.; Scardovi, A.L.; Nieddu, G.; Cerisoli, L.; et al. The MYCN Inhibitor BGA002 Restores the Retinoic Acid Response Leading to Differentiation or Apoptosis by the MTOR Block in MYCN-Amplified Neuroblastoma. J. Exp. Clin. Cancer Res. 2022, 41, 160.
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33.
- Mo, S.P.; Coulson, J.M.; Prior, I.A. RAS Variant Signalling. Biochem. Soc. Trans. 2018, 46, 1325–1332.
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS Isoforms and Mutations in Cancer at a Glance. J. Cell Sci. 2016, 129, 1287–1292.
- Prior, I.A.; Lewis, P.D.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012, 72, 2457–2467.
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974.
- Wang, W.; Fang, G.; Rudolph, J. Ras Inhibition via Direct Ras Binding—Is There a Path Forward? Bioorg. Med. Chem. Lett. 2012, 22, 5766–5776.
- Whyte, D.B.; Kirschmeier, P.; Hockenberry, T.N.; Nunez-Oliva, I.; James, L.; Catino, J.J.; Bishop, W.R.; Pai, J.-K. K- and N-Ras Are Geranylgeranylated in Cells Treated with Farnesyl Protein Transferase Inhibitors. J. Biol. Chem. 1997, 272, 14459–14464.
- Zorde Khvalevsky, E.; Gabai, R.; Rachmut, I.H.; Horwitz, E.; Brunschwig, Z.; Orbach, A.; Shemi, A.; Golan, T.; Domb, A.J.; Yavin, E.; et al. Mutant KRAS Is a Druggable Target for Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20723–20728.
- Golan, T.; Khvalevsky, E.Z.; Hubert, A.; Gabai, R.M.; Hen, N.; Segal, A.; Domb, A.; Harari, G.; David, E.B.; Raskin, S.; et al. RNAi Therapy Targeting KRAS in Combination with Chemotherapy for Locally Advanced Pancreatic Cancer Patients. Oncotarget 2015, 6, 24560–24570.
- Ramot, Y.; Rotkopf, S.; Gabai, R.M.; Zorde Khvalevsky, E.; Muravnik, S.; Marzoli, G.A.; Domb, A.J.; Shemi, A.; Nyska, A. Preclinical Safety Evaluation in Rats of a Polymeric Matrix Containing an SiRNA Drug Used as a Local and Prolonged Delivery System for Pancreatic Cancer Therapy. Toxicol. Pathol. 2016, 44, 856–865.
- Seth, P.P.; Siwkowski, A.; Allerson, C.R.; Vasquez, G.; Lee, S.; Prakash, T.P.; Wancewicz, E.V.; Witchell, D.; Swayze, E.E. Short Antisense Oligonucleotides with Novel 2′–4′ Conformationaly Restricted Nucleoside Analogues Show Improved Potency without Increased Toxicity in Animals. J. Med. Chem. 2009, 52, 10–13.
- Ross, S.J.; Revenko, A.S.; Hanson, L.L.; Ellston, R.; Staniszewska, A.; Whalley, N.; Pandey, S.K.; Revill, M.; Rooney, C.; Buckett, L.K.; et al. Targeting KRAS-Dependent Tumors with AZD4785, a High-Affinity Therapeutic Antisense Oligonucleotide Inhibitor of KRAS. Sci. Transl. Med. 2017, 9, eaal5253.
- Zhong, Z.; Wen, Z.; Darnell, J.E. Stat3: A STAT Family Member Activated by Tyrosine Phosphorylation in Response to Epidermal Growth Factor and Interleukin-6. Science 1994, 264, 95–98.
- Liu, Y.; Li, P.-K.; Li, C.; Lin, J. Inhibition of STAT3 Signaling Blocks the Anti-Apoptotic Activity of IL-6 in Human Liver Cancer Cells. J. Biol. Chem. 2010, 285, 27429–27439.
- Azare, J.; Leslie, K.; Al-Ahmadie, H.; Gerald, W.; Weinreb, P.H.; Violette, S.M.; Bromberg, J. Constitutively Activated Stat3 Induces Tumorigenesis and Enhances Cell Motility of Prostate Epithelial Cells through Integrin Β6. Mol. Cell. Biol. 2007, 27, 4444–4453.
- Masuda, M.; Suzui, M.; Yasumatu, R.; Nakashima, T.; Kuratomi, Y.; Azuma, K.; Tomita, K.; Komiyama, S.; Weinstein, I.B. Constitutive Activation of Signal Transducers and Activators of Transcription 3 Correlates with Cyclin D1 Overexpression and May Provide a Novel Prognostic Marker in Head and Neck Squamous Cell Carcinoma. Cancer Res. 2002, 62, 3351–3355.
- Danoch, H.; Kalechman, Y.; Albeck, M.; Longo, D.L.; Sredni, B. Sensitizing B- and T- Cell Lymphoma Cells to Paclitaxel/Abraxane—Induced Death by AS101 via Inhibition of the VLA-4—IL10—Survivin Axis. Mol. Cancer Res. 2015, 13, 411–422.
- Ryu, K.; Susa, M.; Choy, E.; Yang, C.; Hornicek, F.J.; Mankin, H.J.; Duan, Z. Oleanane Triterpenoid CDDO-Me Induces Apoptosis in Multidrug Resistant Osteosarcoma Cells through Inhibition of Stat3 Pathway. BMC Cancer 2010, 10, 187.
- Fossey, S.L.; Bear, M.D.; Lin, J.; Li, C.; Schwartz, E.B.; Li, P.-K.; Fuchs, J.R.; Fenger, J.; Kisseberth, W.C.; London, C.A. The Novel Curcumin Analog FLLL32 Decreases STAT3 DNA Binding Activity and Expression, and Induces Apoptosis in Osteosarcoma Cell Lines. BMC Cancer 2011, 11, 112.
- Reilley, M.J.; McCoon, P.; Cook, C.; Lyne, P.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; et al. STAT3 Antisense Oligonucleotide AZD9150 in a Subset of Patients with Heavily Pretreated Lymphoma: Results of a Phase 1b Trial. J. Immunother. Cancer 2018, 6, 119.
- Kortylewski, M.; Swiderski, P.; Herrmann, A.; Wang, L.; Kowolik, C.; Kujawski, M.; Lee, H.; Scuto, A.; Liu, Y.; Yang, C.; et al. In Vivo Delivery of SiRNA to Immune Cells by Conjugation to a TLR9 Agonist Enhances Antitumor Immune Responses. Nat. Biotechnol. 2009, 27, 925–932.
- Tsujimoto, Y.; Finger, L.R.; Yunis, J.; Nowell, P.C.; Croce, C.M. Cloning of the Chromosome Breakpoint of Neoplastic B Cells with the t(14;18) Chromosome Translocation. Science 1984, 226, 1097–1099.
- Vaux, D.L.; Cory, S.; Adams, J.M. Bcl-2 Gene Promotes Haemopoietic Cell Survival and Cooperates with c-Myc to Immortalize Pre-B Cells. Nature 1988, 335, 440–442.
- García-Aranda, M.; Pérez-Ruiz, E.; Redondo, M. Bcl-2 Inhibition to Overcome Resistance to Chemo- and Immunotherapy. Int. J. Mol. Sci. 2018, 19, 3950.
- Cotter, F.E.; Johnson, P.; Hall, P.; Pocock, C.; al Mahdi, N.; Cowell, J.K.; Morgan, G. Antisense Oligonucleotides Suppress B-Cell Lymphoma Growth in a SCID-Hu Mouse Model. Oncogene 1994, 9, 3049–3055.
- Raynaud, F.I.; Orr, R.M.; Goddard, P.M.; Lacey, H.A.; Lancashire, H.; Judson, I.R.; Beck, T.; Bryan, B.; Cotter, F.E. Pharmacokinetics of G3139, a Phosphorothioate Oligodeoxynucleotide Antisense to Bcl-2, after Intravenous Administration or Continuous Subcutaneous Infusion to Mice. J. Pharmacol. Exp. Ther. 1997, 281, 420–427.
- Gagliardi, M.; Ashizawa, A.T. Making Sense of Antisense Oligonucleotide Therapeutics Targeting Bcl-2. Pharmaceutics 2022, 14, 97.
- Ebrahim, A.S.; Kandouz, M.; Liddane, A.; Sabbagh, H.; Hou, Y.; Li, C.; Al-Katib, A. PNT2258, a Novel Deoxyribonucleic Acid Inhibitor, Induces Cell Cycle Arrest and Apoptosis via a Distinct Mechanism of Action: A New Class of Drug for Non-Hodgkin’s Lymphoma. Oncotarget 2016, 7, 42374–42384.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Entry Collection:
Biopharmaceuticals Technology
Revisions:
2 times
(View History)
Update Date:
29 Jul 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No