Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Xiang-Ping Chu | -- | 1853 | 2022-07-28 15:49:15 | | | |
| 2 | Camila Xu | Meta information modification | 1853 | 2022-07-29 03:11:50 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Singh, S.; Yang, F.; Sivils, A.; Cegielski, V.; Chu, X. Regulation of Amylin and Secretases on Alzheimer’s Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/25619 (accessed on 24 July 2026).
Singh S, Yang F, Sivils A, Cegielski V, Chu X. Regulation of Amylin and Secretases on Alzheimer’s Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/25619. Accessed July 24, 2026.
Singh, Som, Felix Yang, Andy Sivils, Victoria Cegielski, Xiang-Ping Chu. "Regulation of Amylin and Secretases on Alzheimer’s Disease" Encyclopedia, https://encyclopedia.pub/entry/25619 (accessed July 24, 2026).
Singh, S., Yang, F., Sivils, A., Cegielski, V., & Chu, X. (2022, July 28). Regulation of Amylin and Secretases on Alzheimer’s Disease. In Encyclopedia. https://encyclopedia.pub/entry/25619
Singh, Som, et al. "Regulation of Amylin and Secretases on Alzheimer’s Disease." Encyclopedia. Web. 28 July, 2022.
Copy Citation
Alzheimer’s disease remains a prevailing neurodegenerative condition which has an array physical, emotional, and financial consequences to patients and society. Among these biomolecules, there are four modulatory mechanisms of interest: alpha-, beta-, gamma-secretases, and amylin. Thus, regulation of these might have a potential therapeutic function for treatment of AD.
amylin
biomolecules
Alzheimer’s disease
1. Introduction
Across the globe, Alzheimer’s disease (AD) is a prevailing cause of dementia and death in elderly populations [1][2]. Clinically, it presents in three stages of cognition sequencing from normal ability to functional impairment and memory loss, colloquially known as dementia [3]. However, recognizable pathologies of AD can be seen up to twenty years prior to onset of clinical symptoms [4]. One of the earliest pathologic hallmarks is accumulation of extracellular amyloid β (Aβ) in the cerebrum [5]. Additionally, neurofibrillary tangles composed of hyperphosphorylated tau are another core histopathologic feature of AD [6][7]. These protein deposits have thus served as major research concentrations for understanding the pathophysiology of this disease.
2. Alpha Secretase Activators
Alpha secretase completes proteolysis of APP which leads down the non-amyloidogenic pathway, where beta secretase completes proteolysis of APP through the amyloidogenic pathway (Figure 1) [8]. Evidence suggests that α-secretase activity is modulated by metalloprotease inhibitors and metal ions, in fact three members of the ADAM (a disintegrin and metalloprotease) family are reported to be candidate α-secretases [8]. ADAMs are type I transmembrane proteins which will be proteolytically active if they have the relevant catalytically active domain [9].
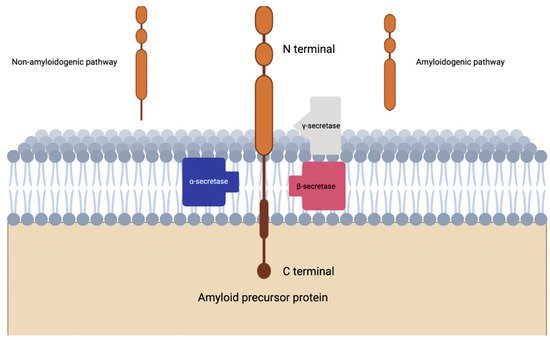
Figure 1. Visualization of amyloidogenic and non-amyloidogenic pathways in the cleavage of amyloid precursor protein. Notice the intramembranous piecing produced by the non-amyloidogenic pathway.
Specifically, investigations have revealed that ADAM10 is the α-secretase that mediates the non-amyloidogenic pathway primarily, while APP cleavage by the β-secretase BACE1 or γ-secretase complex yields the pathogenic Aβ peptide [9]. However, another α-secretase is implicated in the pathology of AD, ADAM17 [10]. A genomic investigation showed that there was a single rare nonsynonymous variant in ADAM17 that co-segregated with an autosomal-dominant pattern of late onset AD in one family [10]. Furthermore, data suggested a strong negative correlation between APP gene expression and ADAM17 in the human brain [10]. Which is further captured by the fact that a p.R215l mutation of ADAM17 led to elevated Aβ formation in vitro [10].
Some investigations born from the idea that α-secretase activation could be therapeutic in AD treatment have been fruitful. In fact, one modifier of α-secretase via the retinoic acid pathway is the vitamin A derivative retinoic acid [11]. The idea here is that retinoic acid activates ADAM10 and thus reduces the amount of Aβ peptide [11]. Evidence shows that vitamin A deficiency leads to an increase in Aβ peptide levels in wild-type (WT) mice [11]. Even more interesting, the rescue of this deficiency has led to increased non-amyloidogenic processing [11].
As an extension of this idea, some researchers looked at the peroxisome proliferator-activated receptor (PPAR-α) which is another component of the retinoic acid pathway. PPAR-α has been demonstrated to activate ADAM10 transcription, eventually leading to a reduced production of pathogenic Aβ peptides [11]. However, there have been three clinical trials related to vitamin A supplementation in AD and only one of them had positive findings—which included only a maintained baseline cognitive performance over 12 months without any improvement [12].
However, there is some promise in utilizing the lipid-lowering medication gemfibrozil which activates PPAR-α and was shown to both inhibit the production of Aβ via upregulation of ADAM10 and stimulate cellular clearance by inducing lysosomal biogenesis in the 5XFAD transgenic model of AD [13][14][15][16][17]. In a phase II parallel-design, double-blind, placebo-controlled trial targeting predementia AD, there was a significant decline on the CANS-MCI cognitive battery, and almost statistically significant declines in Aβ 42 levels and hippocampal atrophy [18]. These findings tentatively validate the pathophysiologic hypothesis underpinning α-secretases as treatment or prevention for AD, but it is fair to say the evidence is less than convincing that these may be formidable treatments.
3. Beta-Secretase Inhibitors
Another transmembrane aspartic protease is β-secretase (BACE1 and BACE2), which serves as another enzyme that is implicated in the aggregation of Aβ plaque [19]. In support of the Aβ amyloid hypothesis for AD pathogenesis, BACE1 is theorized to cleave APP into Aβ plaques within Golgi endosomes and lysosomes, thus making the enzyme a potential modulation site to reduce neurodegenerative effects of Aβ plaques [20]. BACE1 has a crystalline structure with different structural conformations dependent on its activation/inactivation [19]. More specifically, BACE1 activity is highly associated with the flexibility of the flap covering the active binding site of the protease [21]. The importance of BACE1 has been seen in several gene knock-out (KO) studies done by researchers in attempts to observe any reduction of Aβ plaque with gene deletions. One such study demonstrated that partial BACE1 gene deletion (50% to 70%) induces synaptic plasticity deficit in adult mice [22]. Similarly, mouse-models with BACE1 gene KO alterations reported less Aβ build up in cerebral tissue [23]. While these evidence supports the idea that inhibition of the β-secretase protein has positive downstream effects in reducing Aβ plaque, complete lack of the BACE1 has also been shown to have putative detrimental effects to normal neurophysiology [24]. Although BACE1 gene was first identified through its critical role in AD pathogenesis, it is also a vital contributor of muscle spindle fiber formation and maturation [24]. Without the protein expressed in KO mice, researchers found unnatural alteration in mice movement due to impaired muscle proprioception and coordination [24].
The majority of β-secretase inhibitor development and testing surrounds the inhibition of BACE1. Its close homologue, BACE2, is not highly-expressed in brain tissue and is instead found at higher concentration in pancreatic islet cells and a variety of other peripheral tissues [25]. Due to this, selective BACE2 inhibitors developed by researchers primarily concern the treatment of type 2 diabetes mellitus (T2DM) and but not AD [26]. However, several strides have been made to establish BACE2′s role as a potential contributor to AD risk despite not having significant documentation of its function within the central nervous system (CNS). Recent findings identified BACE2 to have conditional β-secretase activity that is dependent on a mutation in the juxtamembrane helix domain of the protein [27]. These results suggest a potential AD therapeutic that targets BACE2 gene without the adverse effects observed in inhibition of BACE1 gene [28]. Considering the expression sites of BACE2 gene, data suggests that it is possible for dual pharmacological targeting of the β-secretase for both T2DM and AD [27]. BACE2 gene may also alternatively act as a Aβ protease in addition to its better-understood function as an APP protease [29]. Overexpression of BACE2 gene may also have anti-amyloidogenic effects as suggested by Sun et al. [30]. It is important to note that cross-reactivity between BACE1 and BACE2 exists, and thus, therapeutics targeting BACE1 may also have unpredicted effects in BACE2 pleiotropism for better or for worse [29][30][31][32].
4. Gamma-Secretase Inhibitors
In line with targeting causes of the pathological accumulation of Aβ plaque in AD progression, the protease γ-secretase serves as another spotlight for therapeutic research within the field. This protein has four subunits that piece together to function in cleaving the transmembrane domains of over 100 membrane proteins [33]. In the amyloidogenic pathway of the amyloid cascade hypothesis, it was thought that γ-secretase was the final step before Aβ plaque formation, cleaving intramembrane segments on the fragment created by the β-secretase enzyme (also see Figure 1) [34]. Attempts at exploiting the role of γ-secretase in AD were trialed under the therapeutic efforts of γ-secretase inhibitors. Subsequently, the discoveries and contributions of this drug class will serve as the focus for this section.
The four subunits composing γ-secretase include presenilin (Psn), nicastrin (Nct), anterior- pharynx-defective-1 (Aph-1), and presenilin enhancer-2 (Pen-2) [35]. In the broad scheme of AD, presenilin proteins are of particular pathophysiologic importance due to dominant mutations that cause increased accumulation of Aβ42 and Aβ40 [36], consequently contributing to early-onset familial AD [37]. With regard to γ-secretase, the presenilin protein plays an integral role in enzymatic function as it is the catalyst for the function of this protease complex [34]. As a multi-transmembrane protein in itself, Psn forms its own molecular mass complex that is easily degraded [38]. However, when this holoprotein Psn is stabilized by Aph-1 and Nct, it forms a stable high molecular mass complex that prevents its degradation yet still renders the complex enzymatically inactive. Only when Pen-2 lyses the high molecular mass complex and presents the active aspartate residues of Psn does the structure come together as the active γ-secretase enzyme [38]. It is important to note that due to the multiplex nature of the γ-secretase complex, this protein is able to perform an array of different functions in unalike cells [39].
γ-secretase is unspecific and has many lipid-based intramembrane substrates that it targets, particularly among type I transmembrane proteins. APP was the first substrate demonstrated to be targeted by γ-secretase [39]. γ-secretase targets the c-terminal of the Aβ domain of APP after upstream processing by α- and β-secretases, releasing Aβ and APP intracellular domain (AICD) [40]. While the role of AICD in signal transduction is controversial [41], there is evidence that AICD is produced by the action of γ-secretase at the plasma membrane/early endosome location of cells [42]. Thus, while scientist is still unsure whether this plays a role in active disease pathophysiology or whether it is a downstream effect of AD, knowing where γ-secretase works helps to localize exactly where enzyme action occurs in cells and as a result, helps to determine where amyloid is formed.
5. Amylin Agonists
Faced with data that challenges the AH, researchers have reassessed the function of amylin, also known as islet amyloid polypeptide, in the pathology of AD. Human amylin was first found in the pancreas, where it is co-secreted with insulin from pancreatic B cells [43]. Endocrinology research discovered that in the early stages of Type II diabetes mellitus, amylin levels are higher than usual. Additionally, this protein is prone to misfold and then form oligomers and fibrils when it is without a matched amount of stabilizing insulin [44][45]. Taking things a step further, data have shown that type 2 diabetes mellitus is a major risk factor for the development of AD [46]. Research has actually shown that these peptides cause death of neurons via induction of proapoptotic genes in a mechanistically similar way as Aβ plaques [47]. Outside of pathology, amylin reduces food intake and body weight, in addition to modulating nociception and cognitive function [47].
These findings paint the picture that amylin, as with Aβ peptides, are cytotoxic to neurons and pathogenic leading to AD. However, some findings suggest otherwise. For example, pramlintide—a synthetic amylin analogue—has been reported to attenuate both Aβ and amylin induced depression of LTP in the hippocampus of AD mice [48]. A noted amylin receptor antagonist (AC253) produces the same attenuation [47]. This apparent contrast puzzles researchers and has led to at least one group initially hypothesizing that there may be ‘biased agonism’, as has been recently reported for the calcitonin receptor (CTR) component of the amylin receptor (AMY). Regardless of the mechanism in question, follow-up experiments involving the administration of amylin and pramlintide in transgenic mouse models of AD lead to improvement in behavioral measures and an efflux of brain Aβ [49]. It is theorized that the excess amylin may act as a ‘peripheral sink’ which leads to the exodus of amyloid across the BBB in addition to amylin receptor interaction [47].
References
- Dumurgier, J.; Sabia, S. Epidemiology of Alzheimer’s disease: Latest trends. Rev. Prat. 2020, 70, 149–151.
- Zhang, X.-X.; Tian, Y.; Wang, Z.-T.; Ma, Y.-H.; Tan, L.; Yu, J.-T. The Epidemiology of Alzheimer’s Disease Modifiable Risk Factors and Prevention. J. Prev. Alzheimers Dis. 2021, 8, 313–321.
- Vermunt, L.; Sikkes, S.A.M.; van den Hout, A.; Handels, R.; Bos, I.; van der Flier, W.M.; Kern, S.; Ousset, P.-J.; Maruff, P.; Skoog, I.; et al. Duration of Preclinical, Prodromal, and Dementia Stages of Alzheimer’s Disease in Relation to Age, Sex, and APOE Genotype. Alzheimers Dement. J. Alzheimers Assoc. 2019, 15, 888–898.
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s Disease: Definition, Natural History, and Diagnostic Criteria. Alzheimers Dement. J. Alzheimers Assoc. 2016, 12, 292–323.
- Jansen, W.J.; Ossenkoppele, R.; Knol, D.L.; Tijms, B.M.; Scheltens, P.; Verhey, F.R.J.; Visser, P.J.; Amyloid Biomarker Study Group; Aalten, P.; Aarsland, D.; et al. Prevalence of Cerebral Amyloid Pathology in Persons without Dementia: A Meta-Analysis. JAMA 2015, 313, 1924–1938.
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a Biological Definition of Alzheimer’s Disease. Alzheimers Dement. J. Alzheimers Assoc. 2018, 14, 535–562.
- Ferrari, C.; Sorbi, S. The Complexity of Alzheimer’s Disease: An Evolving Puzzle. Physiol. Rev. 2021, 101, 1047–1081.
- Ishiura, S.; Asai, M.; Hattori, C.; Hotoda, N.; Szabo, B.; Sasagawa, N.; Tanuma, S. APP α-Secretase, a Novel Target for Alzheimer Drug Therapy; Landes Bioscience: Austin, TX, USA, 2013.
- Saftig, P.; Lichtenthaler, S.F. The Alpha Secretase ADAM10: A Metalloprotease with Multiple Functions in the Brain. Prog. Neurobiol. 2015, 135, 1–20.
- Hartl, D.; May, P.; Gu, W.; Mayhaus, M.; Pichler, S.; Spaniol, C.; Glaab, E.; Bobbili, D.R.; Antony, P.; Koegelsberger, S.; et al. A Rare Loss-of-Function Variant of ADAM17 Is Associated with Late-Onset Familial Alzheimer Disease. Mol. Psychiatry 2020, 25, 629–639.
- Peron, R.; Vatanabe, I.P.; Manzine, P.R.; Camins, A.; Cominetti, M.R. Alpha-Secretase ADAM10 Regulation: Insights into Alzheimer’s Disease Treatment. Pharm. Basel Switz. 2018, 11, 12.
- Remington, R.; Bechtel, C.; Larsen, D.; Samar, A.; Page, R.; Morrell, C.; Shea, T.B. Maintenance of Cognitive Performance and Mood for Individuals with Alzheimer’s Disease Following Consumption of a Nutraceutical Formulation: A One-Year, Open-Label Study. J. Alzheimers Dis. JAD 2016, 51, 991–995.
- Chandra, S.; Pahan, K. Gemfibrozil, a Lipid-Lowering Drug, Lowers Amyloid Plaque Pathology and Enhances Memory in a Mouse Model of Alzheimer’s Disease via Peroxisome Proliferator-Activated Receptor α. J. Alzheimers Dis. Rep. 2019, 3, 149–168.
- Forner, S.; Kawauchi, S.; Balderrama-Gutierrez, G.; Kramár, E.A.; Matheos, D.P.; Phan, J.; Javonillo, D.I.; Tran, K.M.; Hingco, E.; da Cunha, C.; et al. Systematic Phenotyping and Characterization of the 5xFAD Mouse Model of Alzheimer’s Disease. Sci. Data 2021, 8, 270.
- Jeong, Y.J.; Son, Y.; Park, H.-J.; Oh, S.J.; Choi, J.Y.; Ko, Y.-G.; Lee, H.-J. Therapeutic Effects of Aripiprazole in the 5xFAD Alzheimer’s Disease Mouse Model. Int. J. Mol. Sci. 2021, 22, 9374.
- Oblak, A.L.; Lin, P.B.; Kotredes, K.P.; Pandey, R.S.; Garceau, D.; Williams, H.M.; Uyar, A.; O’Rourke, R.; O’Rourke, S.; Ingraham, C.; et al. Comprehensive Evaluation of the 5XFAD Mouse Model for Preclinical Testing Applications: A MODEL-AD Study. Front. Aging Neurosci. 2021, 13.
- Sáez-Orellana, F.; Octave, J.-N.; Pierrot, N. Alzheimer’s Disease, a Lipid Story: Involvement of Peroxisome Proliferator-Activated Receptor α. Cells 2020, 9, 1215.
- Metabolic and Cerebrovascular Effects of Gemfibrozil Treatment: A Randomized, Placebo-controlled, Double-blind, Phase II Clinical Trial for Dementia Prevention—Jicha—2021—Alzheimer’s & Dementia—Wiley Online Library. Available online: https://alz-journals.onlinelibrary.wiley.com/doi/abs/10.1002/alz.055777 (accessed on 19 May 2022).
- Shimizu, H.; Tosaki, A.; Kaneko, K.; Hisano, T.; Sakurai, T.; Nukina, N. Crystal Structure of an Active Form of BACE1, an Enzyme Responsible for Amyloid β Protein Production. Mol. Cell. Biol. 2008, 28, 3663–3671.
- Lin, X.; Koelsch, G.; Wu, S.; Downs, D.; Dashti, A.; Tang, J. Human Aspartic Protease Memapsin 2 Cleaves the Beta-Secretase Site of Beta-Amyloid Precursor Protein. Proc. Natl. Acad. Sci. USA 2000, 97, 1456–1460.
- Xu, Y.; Li, M.; Greenblatt, H.; Chen, W.; Paz, A.; Dym, O.; Peleg, Y.; Chen, T.; Shen, X.; He, J.; et al. Flexibility of the Flap in the Active Site of BACE1 as Revealed by Crystal Structures and Molecular Dynamics Simulations. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 13–25.
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A Mutation in APP Protects against Alzheimer’s Disease and Age-Related Cognitive Decline. Nature 2012, 488, 96–99.
- Ohno, M.; Sametsky, E.A.; Younkin, L.H.; Oakley, H.; Younkin, S.G.; Citron, M.; Vassar, R.; Disterhoft, J.F. BACE1 Deficiency Rescues Memory Deficits and Cholinergic Dysfunction in a Mouse Model of Alzheimer’s Disease. Neuron 2004, 41, 27–33.
- Cheret, C.; Willem, M.; Fricker, F.R.; Wende, H.; Wulf-Goldenberg, A.; Tahirovic, S.; Nave, K.-A.; Saftig, P.; Haass, C.; Garratt, A.N.; et al. Bace1 and Neuregulin-1 Cooperate to Control Formation and Maintenance of Muscle Spindles. EMBO J. 2013, 32, 2015–2028.
- Voytyuk, I.; Mueller, S.A.; Herber, J.; Snellinx, A.; Moechars, D.; van Loo, G.; Lichtenthaler, S.F.; De Strooper, B. BACE2 Distribution in Major Brain Cell Types and Identification of Novel Substrates. Life Sci. Alliance 2018, 1, e201800026.
- Ghosh, A.K.; Brindisi, M.; Yen, Y.-C.; Lendy, E.K.; Kovela, S.; Cárdenas, E.L.; Reddy, B.S.; Rao, K.V.; Downs, D.; Huang, X.; et al. Highly Selective and Potent Human β-Secretase 2 (BACE2) Inhibitors against Type 2 Diabetes: Design, Synthesis, X-Ray Structure and Structure-Activity Relationship Studies. ChemMedChem 2019, 14, 545–560.
- Wang, Z.; Xu, Q.; Cai, F.; Liu, X.; Wu, Y.; Song, W. BACE2, a Conditional β-Secretase, Contributes to Alzheimer’s Disease Pathogenesis. JCI Insight 2019, 4, 123431.
- Dominguez, D.; Tournoy, J.; Hartmann, D.; Huth, T.; Cryns, K.; Deforce, S.; Serneels, L.; Camacho, I.E.; Marjaux, E.; Craessaerts, K.; et al. Phenotypic and Biochemical Analyses of BACE1- and BACE2-Deficient Mice. J. Biol. Chem. 2005, 280, 30797–30806.
- Huentelman, M.; De Both, M.; Jepsen, W.; Piras, I.S.; Talboom, J.S.; Willeman, M.; Reiman, E.M.; Hardy, J.; Myers, A.J. Common BACE2 Polymorphisms Are Associated with Altered Risk for Alzheimer’s Disease and CSF Amyloid Biomarkers in APOE Ε4 Non-Carriers. Sci. Rep. 2019, 9, 9640.
- Sun, X.; He, G.; Song, W. BACE2, as a Novel APP Theta-Secretase, Is Not Responsible for the Pathogenesis of Alzheimer’s Disease in Down Syndrome. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 1369–1376.
- Vassar, R. BACE1 Inhibition as a Therapeutic Strategy for Alzheimer’s Disease. J. Sport Health Sci. 2016, 5, 388–390.
- Das, B.; Yan, R. A Close Look at BACE1 Inhibitors for Alzheimer’s Disease Treatment. CNS Drugs 2019, 33, 251–263.
- Golde, T.E.; Koo, E.H.; Felsenstein, K.M.; Osborne, B.A.; Miele, L. γ-Secretase Inhibitors and Modulators. Biochim. Biophys. Acta 2013, 1828, 2898–2907.
- Panza, F.; Frisardi, V.; Imbimbo, B.P.; Capurso, C.; Logroscino, G.; Sancarlo, D.; Seripa, D.; Vendemiale, G.; Pilotto, A.; Solfrizzi, V. REVIEW: Γ-Secretase Inhibitors for the Treatment of Alzheimer’s Disease: The Current State. CNS Neurosci. Ther. 2010, 16, 272–284.
- Wolfe, M.S. Inhibition and Modulation of γ-Secretase for Alzheimer’s Disease. Neurotherapeutics 2008, 5, 391–398.
- Lleó, A.; Berezovska, O.; Growdon, J.H.; Hyman, B.T. Clinical, Pathological, and Biochemical Spectrum of Alzheimer Disease Associated with PS-1 Mutations. Am. J. Geriatr. Psychiatry Off. J. Am. Assoc. Geriatr. Psychiatry 2004, 12, 146–156.
- Bentahir, M.; Nyabi, O.; Verhamme, J.; Tolia, A.; Horré, K.; Wiltfang, J.; Esselmann, H.; De Strooper, B. Presenilin Clinical Mutations Can Affect Gamma-Secretase Activity by Different Mechanisms. J. Neurochem. 2006, 96, 732–742.
- Takasugi, N.; Tomita, T.; Hayashi, I.; Tsuruoka, M.; Niimura, M.; Takahashi, Y.; Thinakaran, G.; Iwatsubo, T. The Role of Presenilin Cofactors in the γ-Secretase Complex. Nature 2003, 422, 438–441.
- Liu, X.; Liu, Y.; Ji, S. Secretases Related to Amyloid Precursor Protein Processing. Membranes 2021, 11, 983.
- MacLeod, R.; Hillert, E.-K.; Cameron, R.T.; Baillie, G.S. The Role and Therapeutic Targeting of α-, β- and γ-Secretase in Alzheimer’s Disease. Future Sci. OA 2015, 1, FSO11.
- Müller, T.; Meyer, H.E.; Egensperger, R.; Marcus, K. The Amyloid Precursor Protein Intracellular Domain (AICD) as Modulator of Gene Expression, Apoptosis, and Cytoskeletal Dynamics-Relevance for Alzheimer’s Disease. Prog. Neurobiol. 2008, 85, 393–406.
- Kaether, C.; Schmitt, S.; Willem, M.; Haass, C. Amyloid Precursor Protein and Notch Intracellular Domains Are Generated after Transport of Their Precursors to the Cell Surface. Traffic Cph. Den. 2006, 7, 408–415.
- Westermark, P.; Andersson, A.; Westermark, G.T. Islet Amyloid Polypeptide, Islet Amyloid, and Diabetes Mellitus. Physiol. Rev. 2011, 91, 795–826.
- Johnson, K.H.; O’Brien, T.D.; Jordan, K.; Westermark, P. Impaired Glucose Tolerance Is Associated with Increased Islet Amyloid Polypeptide (IAPP) Immunoreactivity in Pancreatic Beta Cells. Am. J. Pathol. 1989, 135, 245–250.
- Jordan, K.; Murtaugh, M.P.; O’Brien, T.D.; Westermark, P.; Betsholtz, C.; Johnson, K.H. Canine IAPP CDNA Sequence Provides Important Clues Regarding Diabetogenesis and Amyloidogenesis in Type 2 Diabetes. Biochem. Biophys. Res. Commun. 1990, 169, 502–508.
- Mietlicki-Baase, E.G. Amylin in Alzheimer’s Disease: Pathological Peptide or Potential Treatment? Neuropharmacology 2018, 136, 287–297.
- Fu, W.; Patel, A.; Kimura, R.; Soudy, R.; Jhamandas, J.H. Amylin Receptor: A Potential Therapeutic Target for Alzheimer’s Disease. Trends Mol. Med. 2017, 23, 709–720.
- Kimura, R.; MacTavish, D.; Yang, J.; Westaway, D.; Jhamandas, J.H. Pramlintide Antagonizes Beta Amyloid (Aβ)- and Human Amylin-Induced Depression of Hippocampal Long-Term Potentiation. Mol. Neurobiol. 2017, 54, 748–754.
- Zhu, H.; Wang, X.; Wallack, M.; Li, H.; Carreras, I.; Dedeoglu, A.; Hur, J.-Y.; Zheng, H.; Li, H.; Fine, R.; et al. Intraperitoneal Injection of the Pancreatic Peptide Amylin Potently Reduces Behavioral Impairment and Brain Amyloid Pathology in Murine Models of Alzheimer’s Disease. Mol. Psychiatry 2015, 20, 252–262.
More
Information
Subjects:
Pathology; Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Entry Collection:
Neurodegeneration
Revisions:
2 times
(View History)
Update Date:
29 Jul 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No