Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marcel Deckert | -- | 2144 | 2022-07-27 11:48:13 | | | |
| 2 | Mickael Ohanna | + 2 word(s) | 2146 | 2022-07-27 14:39:53 | | | | |
| 3 | Mickael Ohanna | + 1 word(s) | 2147 | 2022-07-27 14:53:29 | | | | |
| 4 | Conner Chen | -4 word(s) | 2143 | 2022-07-28 08:55:50 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ohanna, M.; Biber, P.; Deckert, M.; Ohanna, M. Melanoma and The Ubiquitin Pathway. Encyclopedia. Available online: https://encyclopedia.pub/entry/25580 (accessed on 30 June 2026).
Ohanna M, Biber P, Deckert M, Ohanna M. Melanoma and The Ubiquitin Pathway. Encyclopedia. Available at: https://encyclopedia.pub/entry/25580. Accessed June 30, 2026.
Ohanna, Mickael, Pierric Biber, Marcel Deckert, Mickael Ohanna. "Melanoma and The Ubiquitin Pathway" Encyclopedia, https://encyclopedia.pub/entry/25580 (accessed June 30, 2026).
Ohanna, M., Biber, P., Deckert, M., & Ohanna, M. (2022, July 27). Melanoma and The Ubiquitin Pathway. In Encyclopedia. https://encyclopedia.pub/entry/25580
Ohanna, Mickael, et al. "Melanoma and The Ubiquitin Pathway." Encyclopedia. Web. 27 July, 2022.
Copy Citation
Ubiquitination is a post-translational modification that plays a crucial role in various cellular biological activities and participates in cancer pathogenesis, including melanoma.
melanoma
ubiquitination
invasion
migration
survival
1. Melanoma
Melanoma is a tumor arising from the malignant transformation of melanocytes, pigment-producing cells from the neural crest [1][2]. Several prevalent clinical types of melanoma exist, including cutaneous, uveal, and acral melanoma, according to the location of the transformed melanocyte within the body. The cutaneous melanoma, which is the most dangerous type of skin cancer, accounting for only 4% of skin cancers, but responsible for approximately 80% of deaths. In the top 20 most prevalent cancers overall, the increasing incidence of cutaneous malignant melanoma accounted for around 330,000 cases (1.6%) of all newly diagnosed cancers worldwide and more than 58,000 deaths in 2021 (Global Cancer Observatory (http://gco.iarc.fr/ (accessed on 1 July 2022)). Melanoma development is due to increased genetic and epigenetic alterations, which create an imbalance in homeostatic signaling pathways. This leads to excessive proliferation of out-of-control tumor cells and subsequent dissemination to distant sites, invading organs and metastasizing. This high mortality and morbidity rate is related to its high ability to metastasize, migrate by tropism, and invade specific sites, such as lymph nodes, the brain, lungs, or liver [3][4]. When metastasis is clinically evident, the prognosis is very poor. Consistent with the Breslow thickness and Clark’s model, melanoma progression is generally described as a linear process, passing first horizontally through the epidermis and then vertically with a high level of proliferative capacity as indicated by the Ki-67 index; these are currently the most important prognostic factors in invasive melanoma [5]. The process is recognized today as being much more complex and less linear in nature. At an early stage, patients with primary cutaneous melanomas are managed by surgical excision with a high remission rate [6] and a good long-term prognosis with a high survival rate at 5 years. Furthermore, the difficulty lies in the diagnosis of early forms of melanoma, with initial management primarily guided by the Breslow thickness (BT), which is the depth of tumor invasion into the dermis, a prognostic method that remains unreliable and demonstrates its shortcomings in risk stratification. There is an urgent need to understand the mechanisms and explore methods of making an accurate assessment of small melanomas with metastatic potential in order to target them and develop new treatments.
Over the past 10 years, new therapeutic options have emerged for the management of patients diagnosed with metastatic melanoma with the discovery of an activating mutation in the gene that codes for the protein kinase BRAF, following studies of the mutational “landscape” in melanoma and supported by more recent data from The Cancer Genome Atlas (TCGA) project [7]. About 50% of melanomas carry activating mutations in the BRAF oncogene and about 30% in the NRAS gene leading to overactivation of the mitogen-activated protein kinase (MAPK) pathway, making this signaling cascade a preferential target for melanoma treatment [8]. Several highly selective RAS/MAPK signaling pathway inhibitors have been identified, and this has changed the curative measures applied to these patients. The clinical use of BRAF inhibitors (vemurafenib, dabrafenib, and encorafenib) and MEK inhibitors (trametinib, cobimetinib, and binimetinib) in specific combinations, which have proven to be superior to single-agent therapy, has been shown to be effective in the treatment of melanoma and to significantly improve patients’ progression-free survival and overall survival [9]. Unfortunately, these treatments tend to become progressively less effective, and most patients develop resistance to these inhibitors soon after starting treatment, categorized as acquired resistance, thus adding new categories of patients: those with intrinsic or adaptive (tolerance) resistance to the drugs whose resistance is already present before starting treatment or emerges within hours of treatment. Acquired resistance to inhibitors is commonly caused self-autonomously by genomic rewiring through genetic aberrations of components of the MAPK pathway and its hyperactivation as well as parallel signaling networks such as the PI3K/AKT kinase cascade and alterations in mitochondrial oxidative or redox metabolism [10][11][12][13]. This concept of adaptive capacity is reported in a range of studies as arising from the ability of a sub-population of tumor-derived cells to evolve into a persistent and tolerant state during the initial phases of drug treatment, while most of these tumor cells die [14][15][16]. Single-cell profiling has shown that some of the genetically distinct rare clones, thus fully resistant, can re-enter the cell cycle during treatment and reform the tumor [17][18][19]. The biological events involved are reversible drug or non-genetic adaptation mechanisms that are characterized by changes in the expression of genes involved in cellular plasticity leading to a dedifferentiated state as well as transcriptional, metabolic, and epigenetic signaling pathways [20][21]. Thus, despite the potential of this precision cancer therapy, these treatments also highlight problems of drug resistance that limit the benefit to the patient. Thus, a greater knowledge of the processes of drug adaptation holds the promise of improving the success of melanoma therapy by postponing or reversing acquired resistance.
In recent years, a new era in cancer treatment has emerged with the development of cancer immunotherapy, which seeks to use the patient’s innate and adaptive immune system to recognize and destroy tumor cells. The improved understanding of the immune system based on the modulation of immune checkpoint blocking systems at the cell surface has led to a new age of treatments for melanoma and has become the first-line treatment, even though long-term and durable tumor regression was observed in only a subset of patients [22]. The clinical development of antibodies specifically designed to block immune checkpoint molecules such as CTLA-4 (ipilimumab) and PD-1-/PD-L1-blocking antibodies (pembrolizumab, nivolumab, and atezolizumab) are currently approved as monotherapies for the first-line treatment of advanced melanoma [23][24][25][26]. In spite of such advances, these treatments are also limited by the fact that 40–50% of patients do not respond to these treatments (primary resistance), and, even in responders, resistance to therapy develops in the majority of patients (acquired or secondary resistance). Several causes of resistance to immunotherapy or immune escape have been identified, including defects in antigen presentation in certain tumors or the lack of recognizable foreign antigens. The production of a range of immunosuppressive proteins and metabolic changes both in tumor and T cells are part of the resistance to immune checkpoint blockade therapies [27][28][29].
Accordingly, there is still an unmet need to find alternative therapy options to improve the treatment of melanoma. In this perspective, whether genetic or non-genetic alterations are involved in resistance mechanisms or intrinsic or exogenic drivers leading to epigenetic and transcriptional rearrangement, melanoma cells must fine-tune protein homeostasis and function to support unrestricted cell proliferation.
2. The Ubiquitin Pathway
The modulation of cell signaling depends critically on a repertoire of protein posttranslational modification (PTM) mechanisms, which provide an extra regulatory layer that contributes to the functional diversity of the proteome. Protein ubiquitination has emerged as a modification used by signaling processes to regulate a range of functional behaviors [30][31]. Protein ubiquitination (or ubiquitylation) is the dynamic process of covalent binding of the C-terminal glycine of ubiquitin, a small protein of 76 amino acids, to a lysine moiety on protein substrates, whereby serine, threonine, cysteine, and N-terminal methionine moieties can also be modified [32]. Target proteins can be either monoubiquitinated by the addition of a single ubiquitin molecule or polyubiquitinated by the consecutive addition of several ubiquitins to the previous ubiquitin leading to disparate fates of the modified proteins. The designation of the polyubiquitination chain depends on the type of lysine residue (seven lysine residues: K6, K11, K27, K29, K33, K48, and K63) to which the ubiquitin attaches, which also gives rise to a variety of biological outcomes. Lys48-linked chains mainly tag proteins for 26S proteasome-mediated recognition and degradation while K63-related chains play a variety of non-degradative roles and can alter signaling and transcriptional processes as well as protein interaction or localization [30][31]. Monoubiquitination plays an active role in histone regulation and DNA damage repair, signal transduction, trafficking of receptors, and stress response [33][34]. The ubiquitin–proteasome system (UPS) is implicated in the degradation of more than 80% of short-lived proteins in cells and ensures the elimination of useless, damaged, misfolded, and potentially dangerous proteins and the recycle of ubiquitin. Most of the proteins involved in the cell cycle, cell adhesion, migration, invasion, apoptosis, differentiation, angiogenesis and tumor growth, antigen processing, cytokine signaling, transcription, and DNA damage response are regulated by UPS (Figure 1).
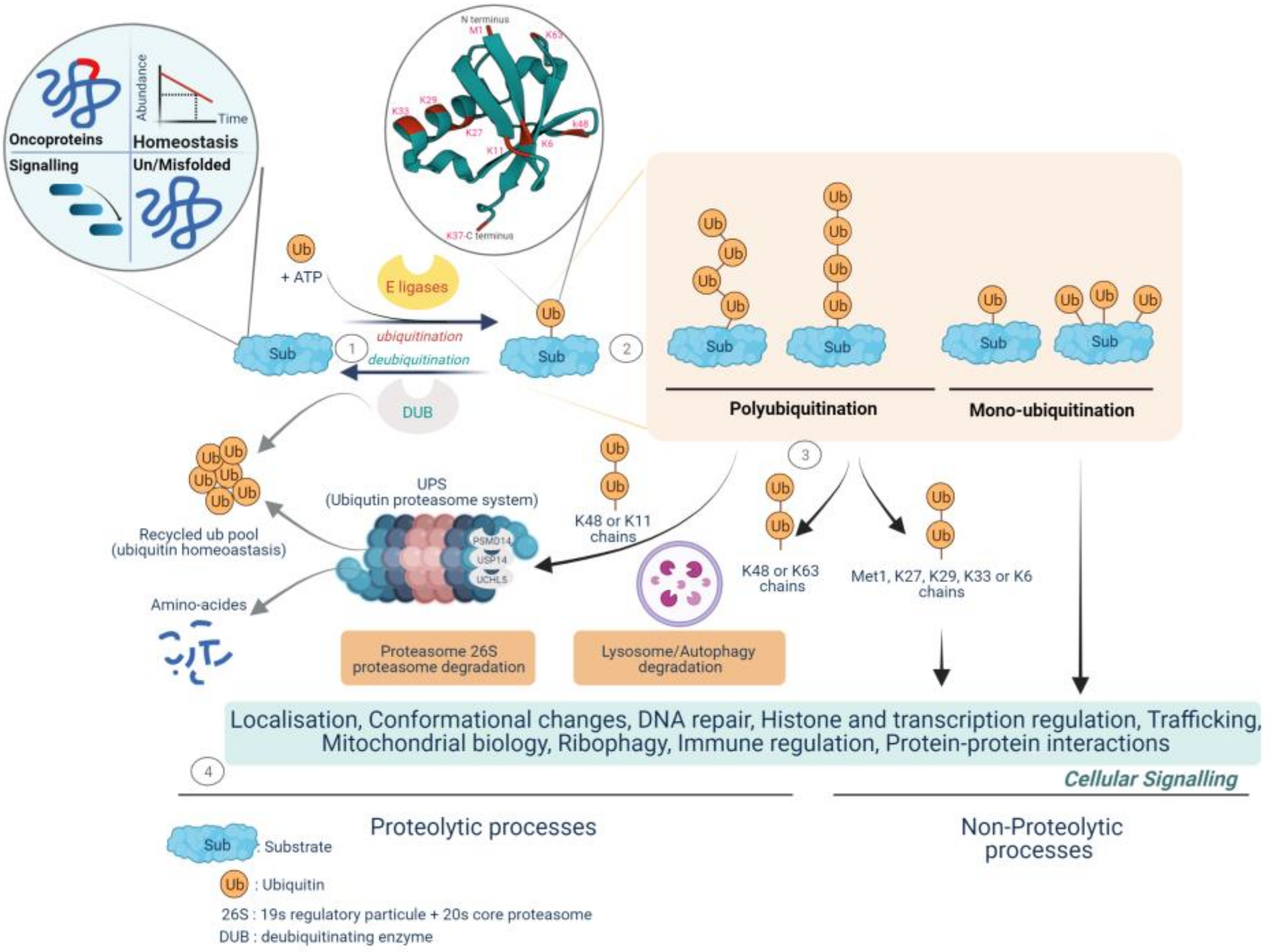
Figure 1. Fate of protein-tethering ubiquitin, a post-translational modification, to proteolytic and non-proteolytic pathways, resulting in specific cellular responses. The ubiquitination process drives protein homeostasis by controlling abundance, temporal and structural integrity, proper localization, and protein non-mutational burden. Called the quality control function, it supports nearly all the cellular functionalities implicated in protein–protein interactions, gene expression, signal transduction cascades, and metabolic pathways. 1—Protein ubiquitination is performed by the coordinated activity of ubiquitin ligase (E1, E2, and E3 enzymes) and deubiquitinating enzymes (DUBs), called deubiquitination, by antagonizing ligase activity and altering the substrate fate. 2—Ubiquitin is covalently transferred (isopeptide bonds) between the C-terminal glycine residue (Gly) and substrate lysine residues (Lys) to form monoubiquitinated proteins or can join up with other ubiquitin molecules at the intrinsic N-terminal Met1 residue and/or at the seven intrinsic Lys residues (Lys6, Lys11, Lys27, Lys29, Lys33, Lys48, and Lys63) and may form multi- or poly-ubiquitin chains. 3—The fate and function of ubiquitinated proteins are affected by the topology and type of ubiquitin- binding. The K48/K11 polyubiquitinated chains have historically been identified as mediating proteasomal degradation of normally folded short-lived proteins and recycling ubiquitin called the ubiquitin–proteasome system (UPS). To date, three deubiquitinases, the metalloprotease PSMD14 and the two cysteine proteases UCHL5 and USP14, have been found to be components of the proteasome 19S regulatory particle implicated in both binding of the ubiquitinated substrate and proteasome activity. 4—The UPS is a selective and irreversible protein removal mechanism that controls signal transduction, cell division, stress response, and immune adaptation. The degradation of misfolded proteins by the UPS or autophagy is mainly mediated through K48/K63 branch chains. Met1/K63/K29 linkage can modulate various non-degradative processes such as signal transduction, DNA repair, and kinase activation. Lys6-linked chains have been identified as being involved in mitophagy. Monoubiquitination plays various roles in such functions as protein trafficking, DNA repair, chromatin remodeling, and regulation of transcription. (Created with BioRender.com, accessed on 20 June 2022).
The classical cascade of ubiquitin conjugation to a protein substrate (ubiquitination) is initiated by a family of ATP-dependent enzymes called E1-activating enzymes, in which ubiquitin is transferred to the cysteine residue of the active site of E1 with an adenylation of the second ubiquitin, subsequently followed by the transfer of the adenylated ubiquitin to the active site of the E2 ubiquitin conjugating-enzymes (E2 conjugators) and completed by the ligation of ubiquitin to the lysine residues of the target proteins by E3 ligases, which plays a key role in the specific type of ubiquitinated substrate and its associated function [35].
Since ubiquitination is a dynamic and reversible process, the removal of ubiquitin is catalyzed by DUBs. Thus, the main function of DUBs, beyond their role in protein stabilization, is to adjust the degree of protein ubiquitination/deubiquitination, protein activity, and subcellular localization, and to preserve the cellular pool of monoubiquitin. Around 100 DUBs are encoded in the human genome and have been divided into six families based on sequence and structure, including UCH (ubiquitin C-terminal hydrolase), USP (ubiquitin-specific protease), OTU (ovarian tumor proteases), Josephin (Machado-Joseph disease, MJD), ZUP1 (zinc finger-containing ubiquitin peptidase), and the JAMMs (JAB1/MPN/Mov34 metalloenzyme) [36][37][38]. The first five families are cysteine proteases, while the JAMM proteins belong to the zinc-dependent metalloproteinase family. There are approximately 57 USPs, 4 UCHs, 15 OTUs, 4 MJDs, 1 ZUP1, and 9 JAMMs (Figure 2). Due to the number and variety of their substrates, DUBs can drive various cellular processes such as the cell cycle, apoptosis, gene transcription, and DNA repair, and they can exhibit versatile functions in tumor progression, such as epithelial-mesenchymal transition (EMT), cancer stem cell development, metastasis, and tumor microenvironment cross-talk. Here, the evidence regarding the involvement of DUBs and related substrates in several biological processes and their relevance in melanoma progression and therapeutic response have been compiled (Figure 3).
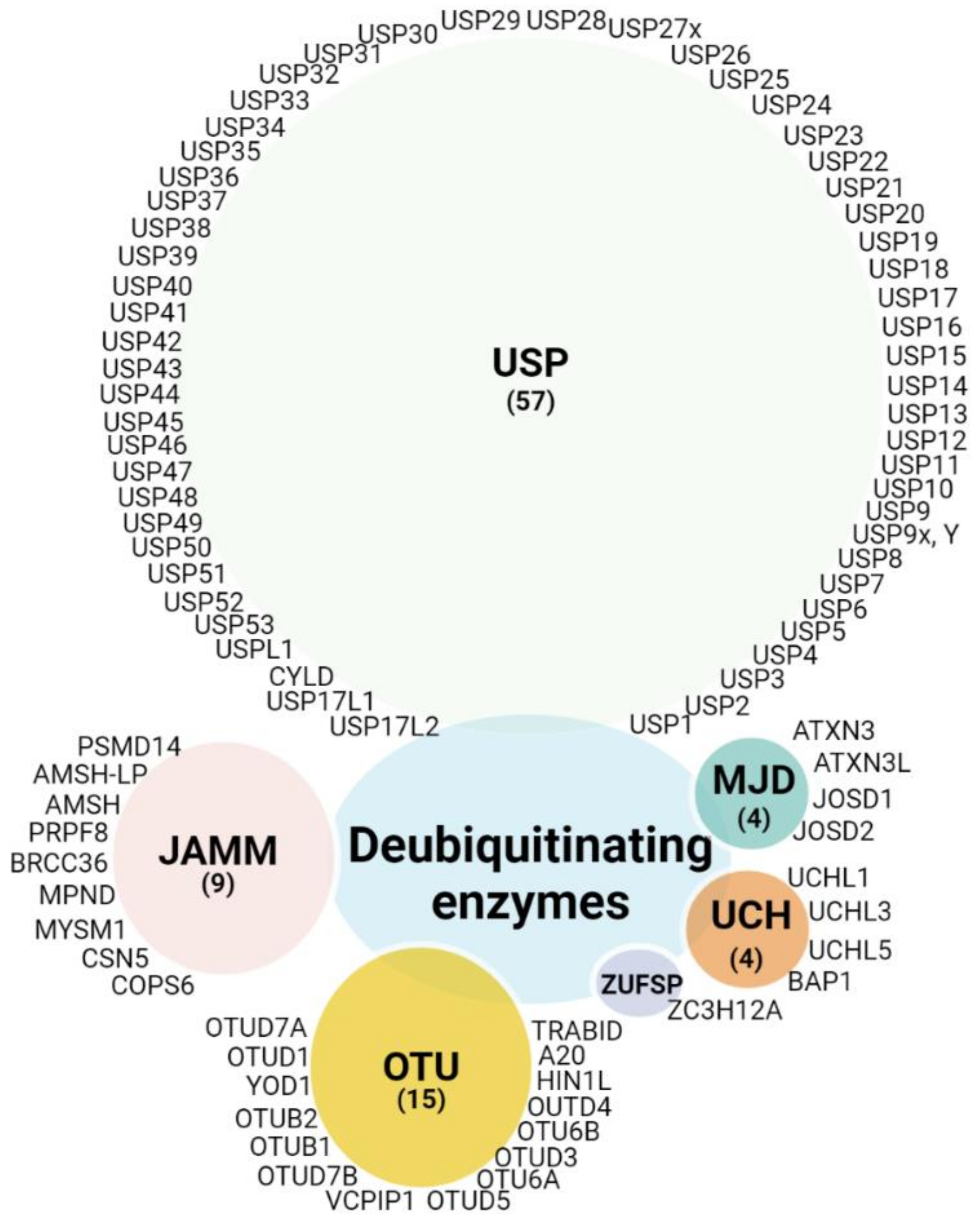
Figure 2. DUB categories: DUBs currently encoded in the human genome are clustered in five classes depicted in different color schemes. Of the six subfamilies, five are cysteine proteases: ubiquitin C-terminal hydrolases (UCH), ubiquitin-specific proteases (USP), zinc finger-containing ubiquitin peptidase (ZUP1), Machado-Joseph disease proteases (MJD, Josephins), and ovarian tumor proteases (OTU), and a one family belongs to the Jab1/Pab1/MPN domain-associated zinc metalloproteases (JAMM). (Created with BioRender.com, (Created with BioRender.com, accessed 20 June 2022).
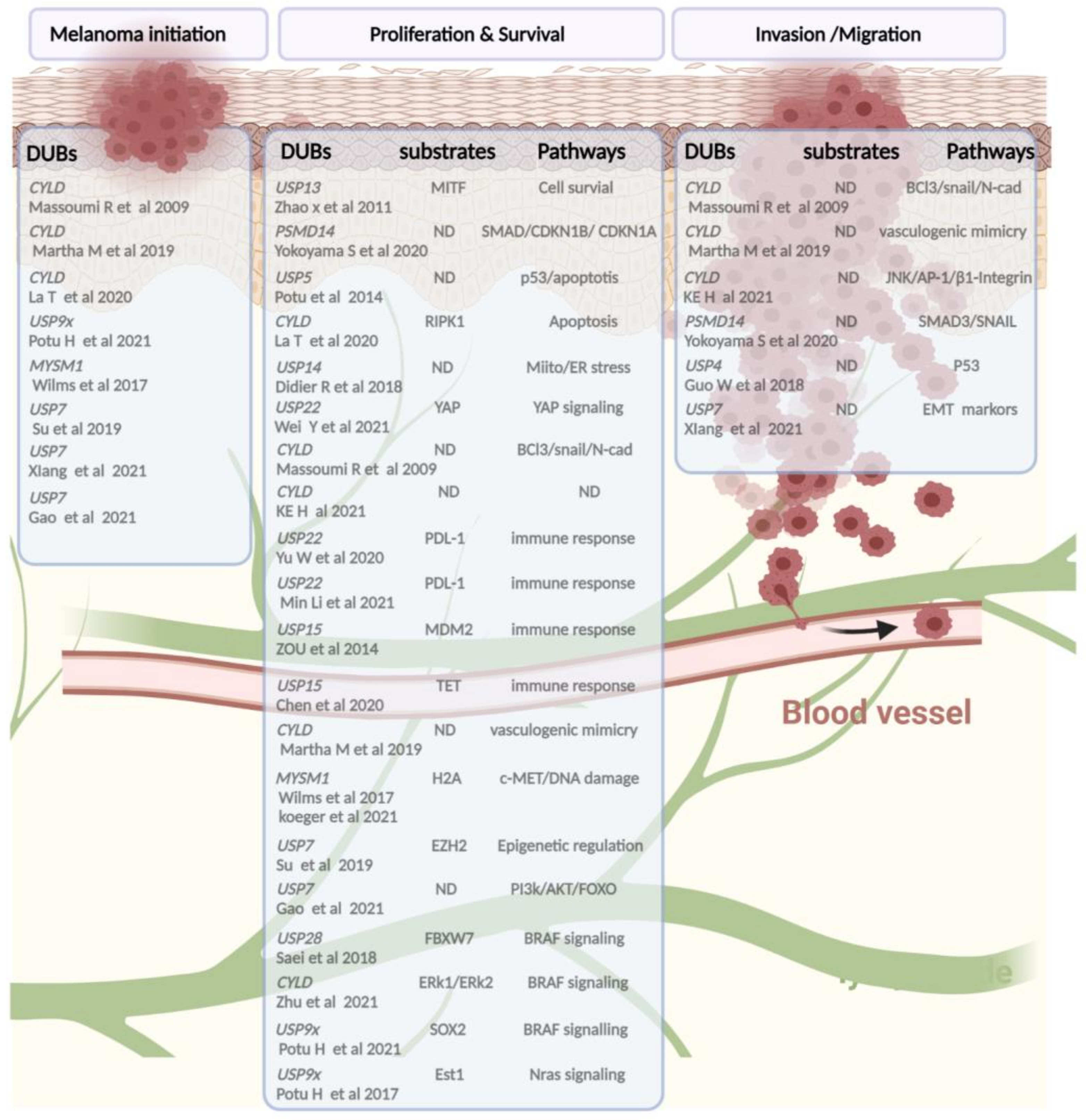
Figure 3. A schematic snapshot of deubiquitinases involved in melanoma pathogenicity based on published studies. From left to right, the following categories are shown: DUBs implied in tumor initiation or progression, mainly found in vivo studies, followed by a list of DUBs and their substrates and/or signaling pathways leading (in bold) to alteration of the proliferation, therapeutic response adaptation, and invasion/migration processes of melanoma cells. (Created with BioRender.com, accessed on 20 June 2022).
References
- Miller, A.J.; Mihm, M.C., Jr. Melanoma. N. Engl. J. Med. 2006, 355, 51–65.
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358.
- Maxwell, R.; Garzon-Muvdi, T.; Lipson, E.J.; Sharfman, W.H.; Bettegowda, C.; Redmond, K.J.; Kleinberg, L.R.; Ye, X.; Lim, M. BRAF-V600 mutational status affects recurrence patterns of melanoma brain metastasis. Int. J. Cancer 2017, 140, 2716–2727.
- Achrol, A.S.; Rennert, R.C.; Anders, C.; Soffietti, R.; Ahluwalia, M.S.; Nayak, L.; Peters, S.; Arvold, N.D.; Harsh, G.R.; Steeg, P.S.; et al. Brain metastases. Nat. Rev. Dis. Primers 2019, 5, 5.
- Pierard-Franchimont, C.; Hermanns-Le, T.; Delvenne, P.; Pierard, G.E. Dormancy of growth-stunted malignant melanoma: Sustainable and smoldering patterns. Oncol. Rev. 2014, 8, 252.
- Balch, C.M.; Gershenwald, J.E.; Soong, S.J.; Thompson, J.F.; Atkins, M.B.; Byrd, D.R.; Buzaid, A.C.; Cochran, A.J.; Coit, D.G.; Ding, S.; et al. Final version of 2009 AJCC melanoma staging and classification. J. Clin. Oncol. 2009, 27, 6199–6206.
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263.
- McArthur, G.A.; Ribas, A. Targeting oncogenic drivers and the immune system in melanoma. J. Clin. Oncol. 2013, 31, 499–506.
- Stadler, S.; Weina, K.; Gebhardt, C.; Utikal, J. New therapeutic options for advanced non-resectable malignant melanoma. Adv. Med. Sci. 2015, 60, 83–88.
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977.
- Wang, L.; Leite de Oliveira, R.; Huijberts, S.; Bosdriesz, E.; Pencheva, N.; Brunen, D.; Bosma, A.; Song, J.Y.; Zevenhoven, J.; Los-de Vries, G.T.; et al. An Acquired Vulnerability of Drug-Resistant Melanoma with Therapeutic Potential. Cell 2018, 173, 1413–1425.e14.
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011, 480, 387–390.
- Moriceau, G.; Hugo, W.; Hong, A.; Shi, H.; Kong, X.; Yu, C.C.; Koya, R.C.; Samatar, A.A.; Khanlou, N.; Braun, J.; et al. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell 2015, 27, 240–256.
- Su, Y.; Ko, M.E.; Cheng, H.; Zhu, R.; Xue, M.; Wang, J.; Lee, J.W.; Frankiw, L.; Xu, A.; Wong, S.; et al. Multi-omic single-cell snapshots reveal multiple independent trajectories to drug tolerance in a melanoma cell line. Nat. Commun. 2020, 11, 2345.
- Torre, E.A.; Arai, E.; Bayatpour, S.; Jiang, C.L.; Beck, L.E.; Emert, B.L.; Shaffer, S.M.; Mellis, I.A.; Fane, M.E.; Alicea, G.M.; et al. Genetic screening for single-cell variability modulators driving therapy resistance. Nat. Genet. 2021, 53, 76–85.
- Emmons, M.F.; Faiao-Flores, F.; Smalley, K.S.M. The role of phenotypic plasticity in the escape of cancer cells from targeted therapy. Biochem. Pharmacol. 2016, 122, 1–9.
- Oren, Y.; Tsabar, M.; Cuoco, M.S.; Amir-Zilberstein, L.; Cabanos, H.F.; Hutter, J.C.; Hu, B.; Thakore, P.I.; Tabaka, M.; Fulco, C.P.; et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature 2021, 596, 576–582.
- Fallahi-Sichani, M.; Becker, V.; Izar, B.; Baker, G.J.; Lin, J.R.; Boswell, S.A.; Shah, P.; Rotem, A.; Garraway, L.A.; Sorger, P.K. Adaptive resistance of melanoma cells to RAF inhibition via reversible induction of a slowly dividing de-differentiated state. Mol. Syst. Biol. 2017, 13, 905.
- Hugo, W.; Shi, H.; Sun, L.; Piva, M.; Song, C.; Kong, X.; Moriceau, G.; Hong, A.; Dahlman, K.B.; Johnson, D.B.; et al. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell 2015, 162, 1271–1285.
- Ohanna, M.; Cerezo, M.; Nottet, N.; Bille, K.; Didier, R.; Beranger, G.; Mograbi, B.; Rocchi, S.; Yvan-Charvet, L.; Ballotti, R.; et al. Pivotal role of NAMPT in the switch of melanoma cells toward an invasive and drug-resistant phenotype. Genes Dev. 2018, 32, 448–461.
- Ratnikov, B.I.; Scott, D.A.; Osterman, A.L.; Smith, J.W.; Ronai, Z.A. Metabolic rewiring in melanoma. Oncogene 2017, 36, 147–157.
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482.
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data from Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2015, 33, 1889–1894.
- Weber, J.S.; D’Angelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H., Jr.; Lao, C.D.; et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015, 16, 375–384.
- Wan, M.T.; Ming, M.E. Nivolumab versus ipilimumab in the treatment of advanced melanoma: A critical appraisal: ORIGINAL ARTICLE: Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 2017, 377, 1345–1356.
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34.
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164.
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355.
- Lim, S.; Phillips, J.B.; Madeira da Silva, L.; Zhou, M.; Fodstad, O.; Owen, L.B.; Tan, M. Interplay between Immune Checkpoint Proteins and Cellular Metabolism. Cancer Res. 2017, 77, 1245–1249.
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229.
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479.
- McClellan, A.J.; Laugesen, S.H.; Ellgaard, L. Cellular functions and molecular mechanisms of non-lysine ubiquitination. Open Biol. 2019, 9, 190147.
- Hicke, L. Protein regulation by monoubiquitin. Nat. Rev. Mol. Cell Biol. 2001, 2, 195–201.
- Hoeller, D.; Dikic, I. Regulation of ubiquitin receptors by coupled monoubiquitination. Subcell. Biochem. 2010, 54, 31–40.
- Mozuraitiene, J.; Gudleviciene, Z.; Vincerzevskiene, I.; Laurinaviciene, A.; Pamedys, J. Expression levels of FBXW7 and MDM2 E3 ubiquitin ligases and their c-Myc and p53 substrates in patients with dysplastic nevi or melanoma. Oncol. Lett. 2021, 21, 37.
- Nijman, S.M.; Luna-Vargas, M.P.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.; Sixma, T.K.; Bernards, R. A genomic and functional inventory of deubiquitinating enzymes. Cell 2005, 123, 773–786.
- Fraile, J.M.; Quesada, V.; Rodriguez, D.; Freije, J.M.; Lopez-Otin, C. Deubiquitinases in cancer: New functions and therapeutic options. Oncogene 2012, 31, 2373–2388.
- Ge, Z.; Leighton, J.S.; Wang, Y.; Peng, X.; Chen, Z.; Chen, H.; Sun, Y.; Yao, F.; Li, J.; Zhang, H.; et al. Integrated Genomic Analysis of the Ubiquitin Pathway across Cancer Types. Cell Rep. 2018, 23, 213–226.e213.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
773
Revisions:
4 times
(View History)
Update Date:
28 Jul 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No