Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Deborah Galpert | -- | 2302 | 2022-07-19 13:14:06 | | | |
| 2 | Peter Tang | Meta information modification | 2302 | 2022-07-20 02:52:20 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Agüero-Chapin, G.; Galpert-Cañizares, D.; Domínguez-Pérez, D.; Marrero-Ponce, Y.; Pérez-Machado, G.; Teijeira, M.; Antunes, A. Non-Classical Peptide Features for Bioactivity Prediction. Encyclopedia. Available online: https://encyclopedia.pub/entry/25278 (accessed on 09 August 2026).
Agüero-Chapin G, Galpert-Cañizares D, Domínguez-Pérez D, Marrero-Ponce Y, Pérez-Machado G, Teijeira M, et al. Non-Classical Peptide Features for Bioactivity Prediction. Encyclopedia. Available at: https://encyclopedia.pub/entry/25278. Accessed August 09, 2026.
Agüero-Chapin, Guillermin, Deborah Galpert-Cañizares, Dany Domínguez-Pérez, Yovani Marrero-Ponce, Gisselle Pérez-Machado, Marta Teijeira, Agostinho Antunes. "Non-Classical Peptide Features for Bioactivity Prediction" Encyclopedia, https://encyclopedia.pub/entry/25278 (accessed August 09, 2026).
Agüero-Chapin, G., Galpert-Cañizares, D., Domínguez-Pérez, D., Marrero-Ponce, Y., Pérez-Machado, G., Teijeira, M., & Antunes, A. (2022, July 19). Non-Classical Peptide Features for Bioactivity Prediction. In Encyclopedia. https://encyclopedia.pub/entry/25278
Agüero-Chapin, Guillermin, et al. "Non-Classical Peptide Features for Bioactivity Prediction." Encyclopedia. Web. 19 July, 2022.
Copy Citation
Artificial intelligence (AI) has been represented by machine learning (ML) algorithms that use sequence-based features for the discovery of new peptidic scaffolds with promising biological activity. From AI perspective, evolutionary algorithms have been also applied to the rational generation of peptide libraries aimed at the optimization/design of antimicrobial peptides (AMPs).
artificial intelligence
machine learning
AMPs
1. Peptide Features Inspired in Molecular Descriptors Used in Cheminformatics
There is a set of chemoinformatics-derived peptide features considered as “non-conventional” because of its in-house development; however, have been successfully applied in the recognition of bioactive peptides by ML-based classifiers [1][2][3][4][5]. The definition of these peptide/protein features is generally inspired on the mathematical formalisms applied to the calculation of molecular descriptors for small organic molecules [6][7], which have been traditionally used to Quantitative-Structure-Activity Relationship (QSAR) studies for drug design/search. Most of them are classified as topological descriptors since they consider the connectivity either between adjacent amino acids (aas) or between aa groups by using both algebraic and statistic invariants [8][9][10].
Those based on algebraic forms express protein/peptide structural topology through the definition of connectivity or adjacency matrices. The elements of these matrices (nij or eij) reflect topological relationships between the aas or aa groups, they are equal to 1 if i and j are adjacent otherwise take the value of 0. Topological indices (TIs) are estimated by applying several algorithms on the connectivity/adjacency matrix. The most common algorithms for the TIs calculation involve the powers of the topological matrix, the multiplication of a property vector by the topological matrix and the multiplication of vector-matrix-vector (Figure 1). Many of the most popular TIs within the cheminformatic have been defined by these algebraic formalisms, such as the Winner index (W) [11], the Randić invariant (χ) [12], Broto–Moreau autocorrelation (ATSd) [13], the Balaban index (J) [14], and the spectral moments introduced by Estrada [6]. Thus, many of them were reformulated to describe the spatial topology of aa sequences at different structural levels, e.g., linear sequences (1D), pseudo-secondary structure (2D) and the 3D-dimensional space [9][10] (Figure 1).
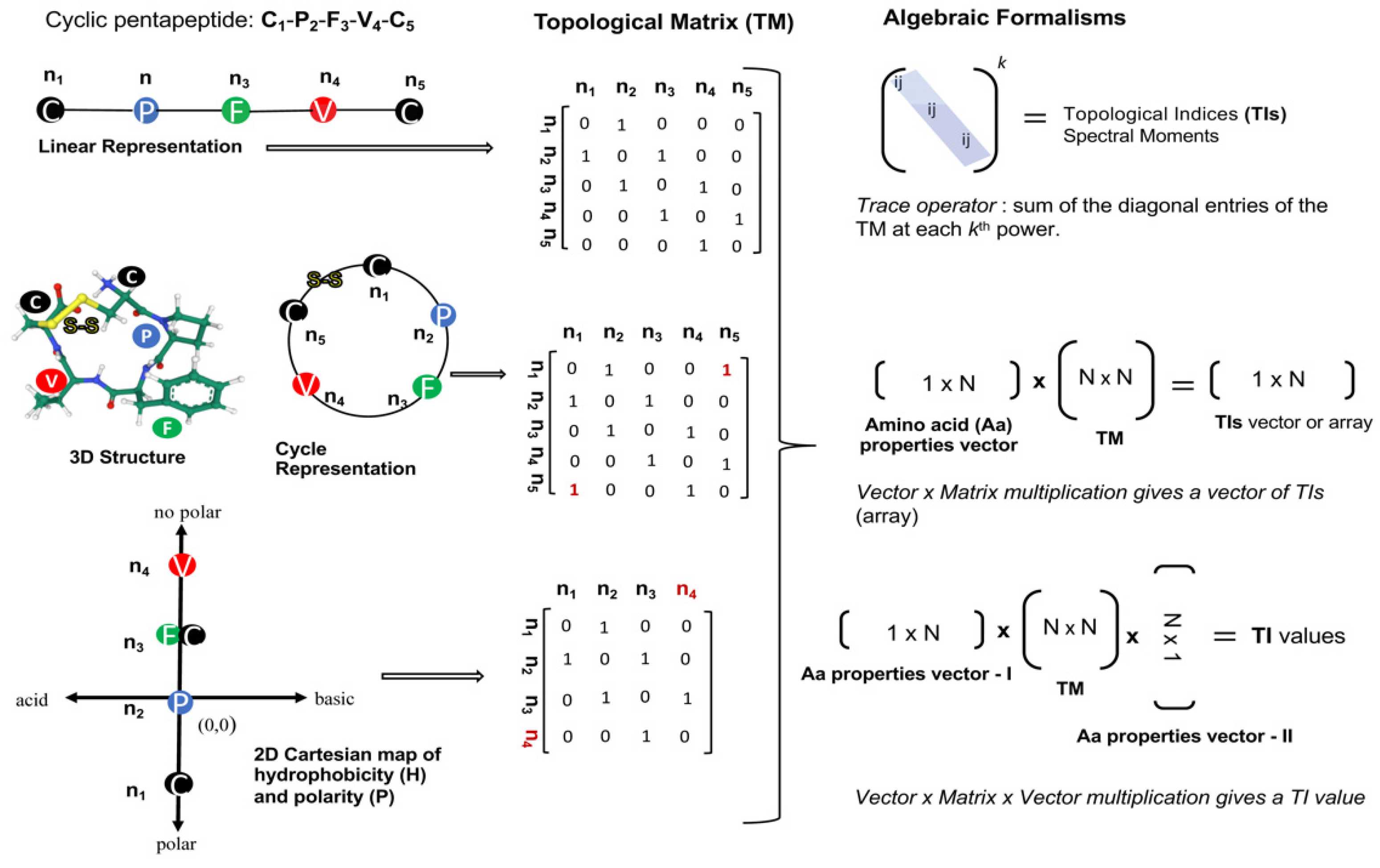
Figure 1. Workflow for the calculation of topological indices from several representation types of the cyclopentapeptide [CPFVC] with promising antiviral activity against the hantavirus cardiopulmonary syndrome [15]. Each peptide representation defines a singular topological matrix (TM) encoding structural features at different degrees. In addition to the several ways to represent the topology of a peptide (linear, circular, 2D-Cartesian), several algebraic formalisms/operators can be applied on the TM to calculate different topological indices (TIs) types. n represents the nodes in the peptidic representations (linear, circular, and Cartesian) as well as in their corresponding TMs, which may contain some elements in red font (e.g., n4 and 1) to highlight differences in structural encoding from the cyclopentapeptide. N indicates the number of rows and columns of matrices involved in TI calculation.
On the other hand, there is another set of topological descriptors that also comes from the chemoinformatic field that have been applied to the identification and design of AMPs [1][3][5][16]. They are not formulated by using algebraic forms but rather they rely on descriptive statistics as invariant operators on the aa properties either along the sequence or the 3D protein structure. In this case, the 1D or 3D topology is encoded by the application of classic cheminformatics algorithms that consider the neighborhood such as autocorrelation [13], Kier-Hall’s electro-topological state [17], Ivanshiuc-Balaban [18], and Gravitational-like operators [19].
1.1. Topological Indices from Algebraic Forms
Among the TIs defined for small molecules, the spectral moments formalism probably is one of the most extended to characterize proteins and peptides structures [9][10][20][21]. The spectral moments may encode peptide structures through the definition of their corresponding topological matrixes and the application of the trace operator on the k-th power of such matrixes (Figure 1).
A sort of stochastic spectral moments applied to the electronic or charge delocalization of the aas within the peptide backbone and the entropy involved on such delocalization, were applied to model the bitter tasting threshold of dipeptides by linear discriminant and regression analyses [4]. These non-standard peptide features provided accuracies higher than 83% in the detection of bitter taste, and the regression models could explain the experimental variance of the bitter tasting threshold in more than 80%. It was shown the non-standard peptide descriptors correlate with the bitter taste as good as or even better than other well-known peptide features like the z-scale [22].
The spectral moments have been also applied to characterize bacteriocins. Bacteriocins are peptidic toxins produced and exported by bacteria as a defense mechanism to kill or inhibit the grow of other strains but the producer. The bacteriocins are very attractive for the development of new antibiotics and anticancer agents, however their high structural diversity represents a challenge for alignment-based predictive tools. Since the hydrophobicity and basicity of bacteriocins are relevant for their antibacterial activity, Agüero-Chapin et al. introduced the 2D-Hydrophobicity and Polarity (2D-HP) maps to pseudo-fold bacteriocin protein sequences in order to derive a set of spectral moments encoding information beyond the linear sequence [23] (Figure 2). These TIs are implemented in the Topological Indices to Biopolymers (TI2BioP) software [24] and were useful to build an AF model based on Linear Discriminant Analysis with a higher sensitivity (66.7%) than the attained by InterProScan (60.2%). In addition, they could detect cryptic bacteriocins, ignored by alignment methods [23].
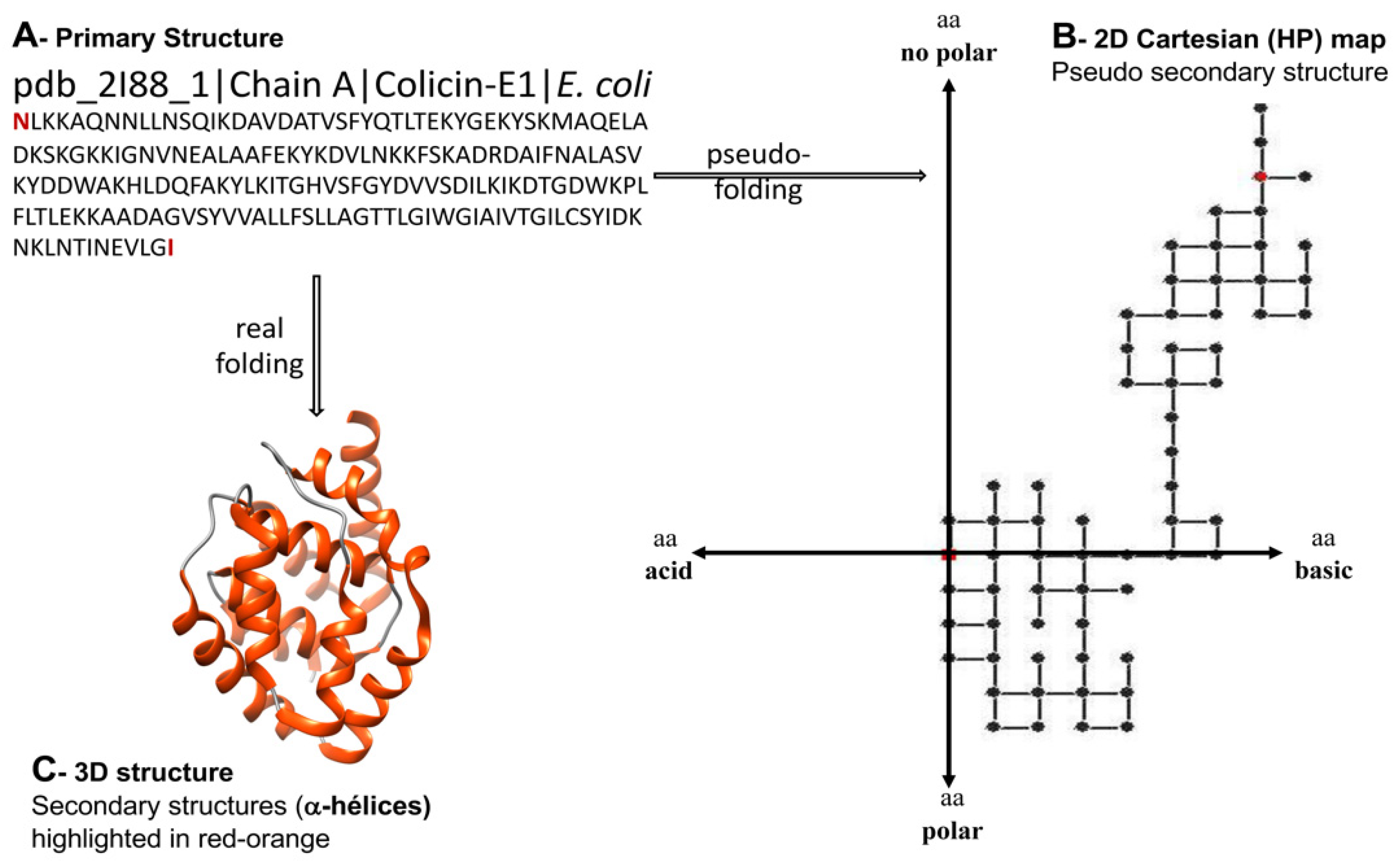
Figure 2. Different structural representations for the channel-forming domain of Colicin E1 (pdb 2I88). A—Primary structure, B—Pseudo secondary Cartesian map of hydrophobicity (H) and polarity (P) (2D Cartesian (HP) map), C—Three-dimensional structure. The 2D Cartesian protein map is an arbitrary bidimensional arrangement (pseudo-folding) of the protein/peptide sequences bearing higher-order useful patterns than contained in linear sequences.
1.2. Topological Indices from Descriptive Statistics
The cheminformatic-derived protein descriptors that have been widely applied to the prediction and design of bioactive peptides were developed and implemented by Ruiz-Blanco et al. in the ProtDCal software [8]. ProtDCal provides a great diversity of protein/peptide descriptors thanks to its divide-and-conquer methodology that considers both the aa properties and those estimated for groups, which can be modified by the neighbourhood through the application of classic previously-mentioned chemoinformatics algorithms. The modified properties of the aas or their resulting groups are later aggregated using statistical operators to estimate local or global descriptors either at sequence or 3D structural level. Although a more detailed description of ProtDCal’s protein descriptors can be found in [8], the Figure 3 shows an schematic representation of the protein descriptor generation process of ProtDCal. The diversity of ProtDCal’s protein descriptors represented by different families stems from combinatorically applying different aa properties, the ways to consider the vicinity to the target aa by several operators, the criteria used to group the aas as well as the invariant operator used for aggregating aa properties within the same array (Figure 3).

Figure 3. Schematic representation of ProtDCal’s descriptors calculation. 1D and 3D protein features implies the application of vicinity operators to modify amino acid (aa) properties while 0D features estimation go straightforward to group the original aa properties according to several grouping criteria.
ProtDCal’s descriptors have been involved in the discovery of antibacterial peptides by developing a non-conventional multi-target QSAR models [3]. Despite the AMPs selected for training were evaluated against multiple targets (Gram-positive bacterial strains), they could be integrated in the same model by modifying their ProtDCal’s descriptors through the Box-Jenkins moving average operator. This operator allows modifying the sequence-based descriptors by subtracting the corresponding mean of the descriptors of all AMPs assayed against the same Gram-positive bacterial strain. This is a way to particularize a sequence-based descriptor by incorporating information about the experimental conditions or biological assays. With this kind of descriptors, the multi-target cheminformatic model displayed percentages of correct classification higher than 90.0% in both training and prediction (test) sets [3].
Similarly, the same authors also applied the Box-Jenkins moving average operator to develop non-conventional multi-task QSAR models able to predict simultaneously antibacterial activity and toxicity [5]. This time, the continuous response variables measured on AMPs such as minimum inhibitory concentration (MIC), cytotoxic concentration at 50% (CC50), and haemolytic concentration at 50% (HC50) were transformed in a binary variable labelled as (1) referred to high antibacterial activity/low cytotoxicity, and (−1) assigned to low antibacterial activity/high cytotoxicity. The ProtDCal’s descriptors that usually encodes only peptide features were modified by the Box-Jenkins moving average operator in order to consider the variability implying the evaluation of the antimicrobial activity and toxicity on different biological systems. Thus, a multi-task QSAR model displayed an accuracy higher than 96% for classifying/predicting peptides was built by using LDA discriminant [5].
ProtDCal’s descriptors have also been involved in the design of new peptides that inhibit the E. coli ATP synthase, as putative antibiotics [16][25]. ProtDCal’s descriptors, implemented in PPI-Detect [26], were applied to predict interactions between peptides and the main subunits of E. coli’s (Ec) and human’s (Hs) F1Fo-ATP synthase. Those peptide with a maximum and a minimum interaction likelihood with EcF1Fo and HsF1Fo were selected for in vitro assays. An overall of three peptides resulted attractive for further optimization steps in the design of new antibiotics [16][25].
More recently, ProtDCal’s protein descriptors were successfully applied to improve the prediction performance of the existing alignment-free models by using the largest experimentally validated non-redundant peptide dataset reported to date, the StarPepDB [27], together with Random Forest (RF) classifiers [1]. Pinacho-Castellanos at al. not only built RF-based models for identifying AMPs, but also addressed the main biological activities reported for them (antibacterial, antifungal, antiparasitic, and antiviral) as endpoints. The specific functions of AMPs were either directly predicted or by a hierarchical classification that first consider the antimicrobial activity. RF-based models, developed with ProtDCal’s descriptors aimed to predict specific activities of AMPs, showed a higher effectivity and reliability than 13 freely available prediction tools. The best reported models were implemented in the AMPDiscover tool [1], publicly available at https://biocom-ampdiscover.cicese.mx/ (accessed on 7 March 2022). Ruiz-Blanco et al. also applied successfully ProtDCal’s descriptors to predict antibacterial peptides by using RF-based models trained with StarPepDB instances and, in a second step they are predicted on what bacterial targets according to their Gram-staining classification could be active by using a multi-classifier. These two RF-based models were implemented in the web server ABP-Finder: https://protdcal.zmb.uni-due.de/ABP-Finder/ (accessed on 7 March 2022) which is freely available but unpublished yet.
2. Integration of Peptide Features from Heterogenous Sources
Considering previous experiences in protein functional classification where protein features from heterogeneous sources have been integrated to improve classification rates; the researchers wonder if this strategy has been applied to peptide classification? In this sense, the integration/combination of alignment-based (AB) and alignment-free (AF) protein features in machine learning models have been evaluated for such purpose. For example, Galpert et al. improved orthologs classification at the twilight zone (<30% of identity) by combining AB and AF protein similarity measures in supervised big data classifiers [28]. It has also been shown that the integration of AB and AF methods gives the best exploration of highly diverse protein classes, such as the nonribosomal peptide synthases (NRPS) represented by their A-domains [29]. Other examples of feature integration methods for remote homology detection can be found in [30], and the one of Borozan’s et al. [31], based on weighted aggregation which is a very inclusive approach avoiding the loss of information.
Regarding AMPs classification improvements by integrating AB and AF peptide features, an algorithm applying AB measures and the SVM algorithm trained with AF pairwise measures was published for increasing AMPs prediction sensitivity [32]. The algorithm consists in two stages. Firstly, AMPs are identified by Basic Local Alignment Search Tool (BLAST) scores, and those peptides that cannot be unequivocally identified by pairwise alignments were inputted in an SVM-based classifier built with AF pairwise similarity scores. The AF similarity scores were estimated with the Lempel–Ziv’s complexity algorithm [33]. The integrative algorithm achieved higher sensitivity performance for AMPs prediction than the prediction tools implemented within the first version of CAMPR3 database [34] and the integrated method proposed by Wang et al. [35]. Wang and colleagues had previously proposed a similar algorithmic workflow where BLAST is used to firstly classify a query peptide against a training set made up by 870 AMPs and 8661 non-AMPs. Classification label is transferred to the query peptide from the matching with highest similarity score. Query peptides that did not match with any within the training set were encoded by protein features like ACC and PseACC and the aas by five of their physicochemical and biochemical properties. As the number of generated features were relatively high, a rigorous feature selection step was performed by applying both the Maximum Relevance, Minimum Redundancy (mRMR) method [36] and the Incremental Feature Selection method [37] before building a Nearest Neighbour (NN)-based predictor. The NN algorithm assign the label AMP or non-AMP to a query peptide according to the class of the nearest neighbour.
Despite the efforts for integrating AB and AF features in a classification peptide system; they have actually been combined through their corresponding algorithms and have not been included in the same model or function. In this sense, AB and AF similarity scores could be combined to build an unique classifier for AMP prediction, as Galpert et al. did it for ortholog detection [28].
3. NMR-Based Features for Peptides
In 2020, the IAMPE webserver (http://cbb1.ut.ac.ir/; accesed on 17 March 2022) was released for an accurate prediction of AMPs by using classical ML-based classifiers trained with both conventional and 13CNMR-based features. The non-conventional 13CNMR-based features for peptides were defined from the quantitative NMR spectra for 13C isotope of the naturally-occurring aas. Firstly, 13CNMR-based features for each aa were calculated using 13CNMR spectra signals. Secondly the aas were grouped according to their 13CNMR-based features by applying Fuzzy c-means clustering algorithm. The resulting aa clusters were used to extract feature vectors along the peptide sequences according to classical “composition”, “transition” and “distribution” patterns. Despite the new information provided by such non-conventional peptide descriptors, authors suggested their combination with physicochemical features to yield higher accuracy for the prediction of active AMP sequences [38].
References
- Pinacho-Castellanos, S.A.; García-Jacas, C.R.; Gilson, M.K.; Brizuela, C.A. Alignment-Free Antimicrobial Peptide Predictors: Improving Performance by a Thorough Analysis of the Largest Available Data Set. J. Chem. Inf. Model. 2021, 61, 3141–3157.
- Agüero-Chapin, G.; Pérez-Machado, G.; Molina-Ruiz, R.; Pérez-Castillo, Y.; Morales-Helguera, A.; Vasconcelos, V.; Antunes, A. TI2BioP: Topological Indices to BioPolymers. Its practical use to unravel cryptic bacteriocin-like domains. Amino Acids 2010, 40, 431–442.
- Speck-Planche, A.; Kleandrova, V.V.; Ruso, J.M.; Cordeiro, M.N.D.S. First Multitarget Chemo-Bioinformatic Model To Enable the Discovery of Antibacterial Peptides against Multiple Gram-Positive Pathogens. J. Chem. Inf. Model. 2016, 56, 588–598.
- De Armas, R.R.; Díaz, H.G.; Molina, R.; González, M.P.; Uriarte, E. Stochastic-based descriptors studying peptides biological properties: Modeling the bitter tasting threshold of dipeptides. Bioorganic Med. Chem. 2004, 12, 4815–4822.
- Kleandrova, V.V.; Ruso, J.M.; Speck-Planche, A.; Cordeiro, M.N.D.S. Enabling the Discovery and Virtual Screening of Potent and Safe Antimicrobial Peptides. Simultaneous Prediction of Antibacterial Activity and Cytotoxicity. ACS Comb. Sci. 2016, 18, 490–498.
- Estrada, E. Spectral Moments of the Edge Adjacency Matrix in Molecular Graphs. 1. Definition and Applications to the Prediction of Physical Properties of Alkanes. J. Chem. Inf. Comput. Sci. 1996, 36, 844–849.
- Mauri, A.; Consonni, V.; Pavan, M.; Todeschini, R. Dragon software: An easy approach to molecular descriptor calculations. Match 2006, 56, 237–248.
- Ruiz-Blanco, Y.B.; Paz, W.; Green, J.; Marrero-Ponce, Y. ProtDCal: A program to compute general-purpose-numerical descriptors for sequences and 3D-structures of proteins. BMC Bioinform. 2015, 16, 162.
- Agüero-Chapin, G.; Molina-Ruiz, R.; Pérez-Machado, G.; Vasconcelos, V.; Rodríguez-Negrin, Z.; Antunes, A. TI2BioP—Topological Indices to BioPolymers. A Graphical–Numerical Approach for Bioinformatics. In Recent Advances in Biopolymers; IntechOpen: Zagreb, Croatia, 2016.
- González-Díaz, H.; González-Díaz, Y.; Santana, L.; Ubeira, F.M.; Uriarte, E.; González-Díaz, H. Proteomics, networks and connectivity indices. Proteomics 2008, 8, 750–778.
- Wiener, H. Structural Determination of Paraffin Boiling Points. J. Am. Chem. Soc. 1947, 69, 17–20.
- Randić, M. Graph theoretical approach to structure-activity studies: Search for optimal antitumor compounds. Prog. Clin. Biol. Res. 1985, 172, 309–318.
- Moreau, G.; Broto, P. The Autocorrelation of a topological structure. A new molecular descriptor. Nouv. J. Chim. 1980, 4, 359–360.
- Balaban, A.T.; Beteringhe, A.; Constantinescu, T.; Filip, P.A.; Ivanciuc, O. Four New Topological Indices Based on the Molecular Path Code. J. Chem. Inf. Model. 2007, 47, 716–731.
- Hall, P.R.; Malone, L.; Sillerud, L.O.; Ye, C.; Hjelle, B.L.; Larson, R.S. Characterization and NMR Solution Structure of a Novel Cyclic Pentapeptide Inhibitor of Pathogenic Hantaviruses. Chem. Biol. Drug Des. 2007, 69, 180–190.
- Ruiz-Blanco, Y.B.; Ávila-Barrientos, L.P.; Hernández-García, E.; Antunes, A.; Agüero-Chapin, G.; García-Hernández, E. Engineering protein fragments via evolutionary and protein–protein interaction algorithms: De novo design of peptide inhibitors for F O F 1 -ATP synthase. FEBS Lett. 2020, 595, 183–194.
- Kier, L.B.; Hall, L.H. An Electrotopological-State Index for Atoms in Molecules. Pharm. Res. 1990, 07, 801–807.
- Ivanciuc, O. Building–Block Computation of the Ivanciuc–Balaban Indices for the Virtual Screening of Combinatorial Libraries. Internet Electron. J. Mol. Des. 2002, 1, 1–9.
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors, 1st ed.; Wiley-VCH: Mannheim, Germany, 2000; Volume 1, p. 667.
- Estrada, E. Characterization of the folding degree of proteins. Bioinformatics 2002, 18, 697–704.
- Estrada, E. Characterization of the amino acid contribution to the folding degree of proteins. Proteins: Struct. Funct. Bioinform. 2004, 54, 727–737.
- Sandberg, M.; Eriksson, L.; Jonsson, J.; Sjöström, A.M.; Wold, S. New Chemical Descriptors Relevant for the Design of Biologically Active Peptides. A Multivariate Characterization of 87 Amino Acids. J. Med. Chem. 1998, 41, 2481–2491.
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120.
- Molina, R.; Agüero-Chapin, G.; Pérez-González, M. TI2BioP (Topological Indices to BioPolymers) Version 2.0; Molecular Simulation and Drug Design (MSDD): Chemical Bioactives Center, Central University of Las Villas, Santa Clara, Cuba, 2011.
- Avila-Barrientos, L.P.; Cofas-Vargas, L.F.; Agüero-Chapin, G.; Hernández-García, E.; Ruiz-Carmona, S.; Valdez-Cruz, N.A.; Trujillo-Roldán, M.; Weber, J.; Ruiz-Blanco, Y.B.; Barril, X.; et al. Computational Design of Inhibitors Targeting the Catalytic β Subunit of Escherichia coli FOF1-ATP Synthase. Antibiotics 2022, 11, 557.
- Romero-Molina, S.; Ruiz-Blanco, Y.B.; Harms, M.; Münch, J.; Sanchez-Garcia, E. PPI-Detect: A support vector machine model for sequence-based prediction of protein-protein interactions. J. Comput. Chem. 2019, 40, 1233–1242.
- Aguilera-Mendoza, L.; Marrero-Ponce, Y.; Beltran, J.A.; Tellez Ibarra, R.; Guillen-Ramirez, H.A.; Brizuela, C.A. Graph-based data integration from bioactive peptide databases of pharmaceutical interest: Toward an organized collection enabling visual network analysis. Bioinformatics 2019, 35, 4739–4747.
- Galpert, D.; Fernández, A.; Herrera, F.; Antunes, A.; Molina-Ruiz, R.; Agüero-Chapin, G. Surveying alignment-free features for Ortholog detection in related yeast proteomes by using supervised big data classifiers. BMC Bioinform. 2018, 19, 166.
- Agüero-Chapin, G.; Molina-Ruiz, R.; Maldonado, E.; de la Riva, G.; Sánchez-Rodríguez, A.; Vasconcelos, V.; Antunes, A. Exploring the adenylation domain repertoire of nonribosomal peptide synthetases using an ensemble of sequence-search methods. PLoS ONE 2013, 8, e65926.
- Agüero-Chapin, G.; Galpert, D.; Molina-Ruiz, R.; Ancede-Gallardo, E.; Pérez-Machado, G.; De la Riva, G.A.; Antunes, A. Graph Theory-Based Sequence Descriptors as Remote Homology Predictors. Biomolecules 2019, 10, 26.
- Borozan, I.; Watt, S.; Ferretti, V. Integrating alignment-based and alignment-free sequence similarity measures for biological sequence classification. Bioinformatics 2015, 31, 1396–1404.
- Ng, X.Y.; Rosdi, B.A.; Shahrudin, S. Prediction of Antimicrobial Peptides Based on Sequence Alignment and Support Vector Machine-Pairwise Algorithm Utilizing LZ-Complexity. BioMed Res. Int. 2015, 2015, 1–13.
- Empel, A.; Ziv, J. On the complexity of finite sequences. IEEE Trans. Inf. Theory 1976, 22, 75–81.
- Waghu, F.H.; Barai, R.S.; Gurung, P.; Idicula-Thomas, S. CAMP R3: A database on sequences, structures and signatures of antimicrobial peptides: Table 1. Nucleic Acids Res. 2016, 44, D1094–D1097. Available online: http://www.ncbi.nlm.nih.gov/pubmed/26467475 (accessed on 23 January 2019).
- Wang, P.; Hu, L.; Liu, G.; Jiang, N.; Chen, X.; Xu, J.; Zheng, W.; Li, L.; Tan, M.; Chen, Z.; et al. Prediction of Antimicrobial Peptides Based on Sequence Alignment and Feature Selection Methods. PLoS ONE 2011, 6, e18476.
- Peng, H.; Long, F.; Ding, C. Feature selection based on mutual information criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 2005, 27, 1226–1238.
- Kohavi, R.; John, G.H. Wrappers for feature subset selection. Artif. Intell. 1997, 97, 273–324.
- Kavousi, K.; Bagheri, M.; Behrouzi, S.; Vafadar, S.; Atanaki, F.F.; Lotfabadi, B.T.; Ariaeenejad, S.; Shockravi, A.; Moosavi-Movahedi, A.A. IAMPE: NMR-Assisted Computational Prediction of Antimicrobial Peptides. J. Chem. Inf. Model. 2020, 60, 4691–4701.
More
Information
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
911
Revisions:
2 times
(View History)
Update Date:
20 Jul 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No