Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rex Devasahayam Arokia Balaya | -- | 1701 | 2022-07-04 10:59:08 | | | |
| 2 | Conner Chen | Meta information modification | 1701 | 2022-07-05 02:46:28 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Rex, D.A.B.; Prasad, T.S.K.; Kandasamy, R.K. Major Cell Death Pathways. Encyclopedia. Available online: https://encyclopedia.pub/entry/24790 (accessed on 23 July 2026).
Rex DAB, Prasad TSK, Kandasamy RK. Major Cell Death Pathways. Encyclopedia. Available at: https://encyclopedia.pub/entry/24790. Accessed July 23, 2026.
Rex, Devasahayam Arokia Balaya, Thottethodi Subrahmanya Keshava Prasad, Richard K. Kandasamy. "Major Cell Death Pathways" Encyclopedia, https://encyclopedia.pub/entry/24790 (accessed July 23, 2026).
Rex, D.A.B., Prasad, T.S.K., & Kandasamy, R.K. (2022, July 04). Major Cell Death Pathways. In Encyclopedia. https://encyclopedia.pub/entry/24790
Rex, Devasahayam Arokia Balaya, et al. "Major Cell Death Pathways." Encyclopedia. Web. 04 July, 2022.
Copy Citation
Programmed cell death (PCD) or apoptosis is an important form of cell-autonomous immune control over intracellular pathogens. PCD is mainly responsible for regulating animal development and tissue homeostasis, which regularly occurs in a broad range of human diseases, including immunological, developmental problems, neurodegeneration, and cancer.
cell death
death receptors
viral infection
1. Major Cell Death Pathways
The Nomenclature Committee on Cell Death (NCCD) has developed guidelines for the morphological, biochemical, and functional definitions and interpretations of cell death. The NCCD’s mission is to provide a widely accepted nomenclature for cell death in order to support the field’s continued development. Various programs of RCD were described, and their research continues to progress. Accordingly, it defined terms based on biological functions such as intrinsic apoptosis, extrinsic apoptosis, and MPT-driven necrosis and pyroptosis from a molecular perspective [1][2][3] (Figure 1).
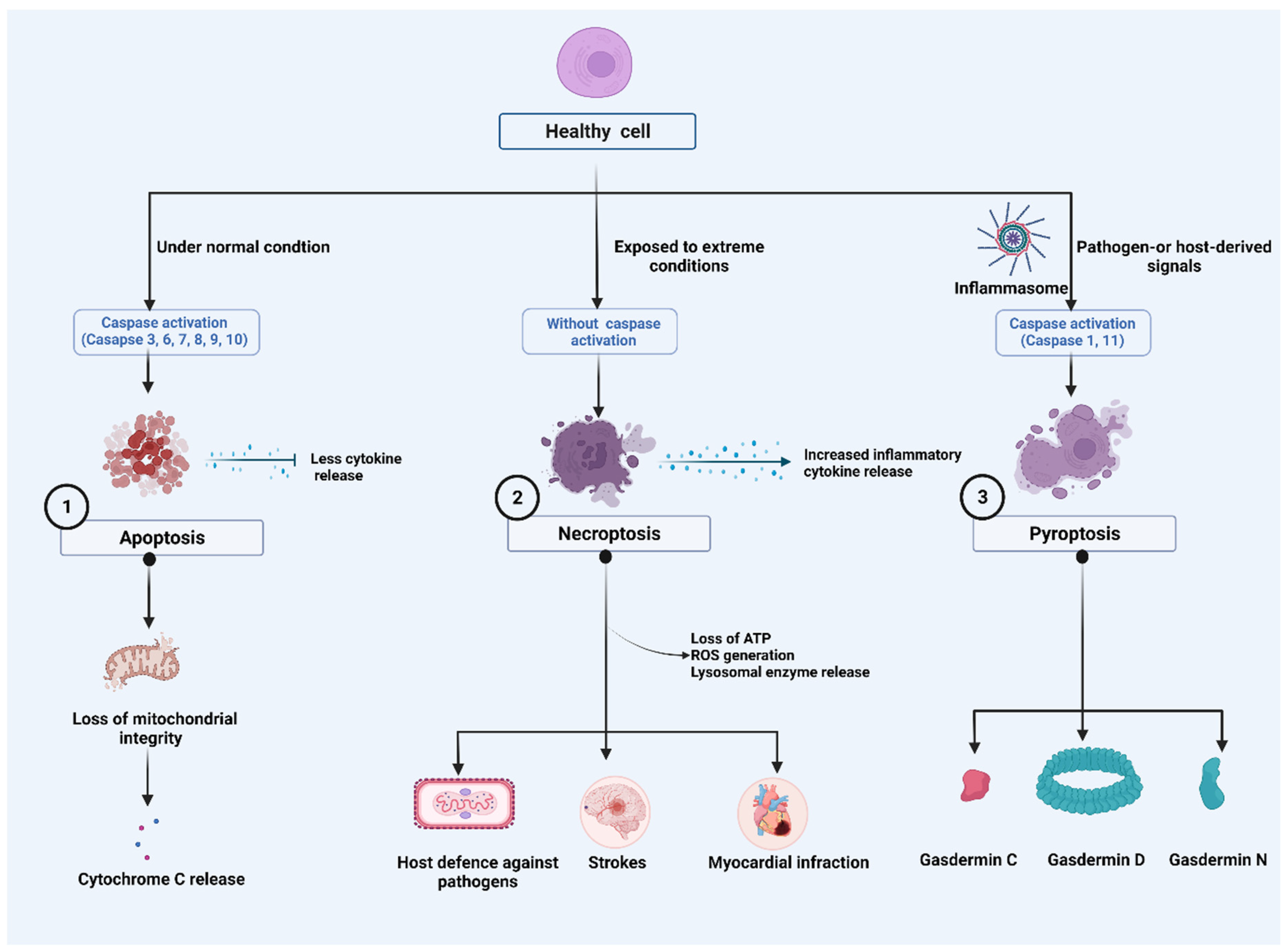
Figure 1. Major cell death pathways: (1). Harmless apoptosis: Mitochondria play a central role in the initiation of apoptosis. During early phases of cell death, cytochrome c (Cyt c) is often released from mitochondria. (2). A pathogenic mediator, necroptosis, plays a role in a variety of diseases and in the fight against viral infection. Despite having caspase activation, necroptosis plays a role in triggering and amplifying inflammation. Inflammatory responses are amplified by the activity of cytokines, which activate pro-inflammatory genes and lead to regulated cell death. Stress, infection, and injury can initiate the response by inducing regulated cell death directly as well as activating immune cells and cytokines. Receptor interacting protein-mediated reactive oxygen species (ROS) production, loss of ATP and lysosomal release can reactivate necroptosis, forming a positive feedback loop mechanism. (3). Orchestration of NLR family pyrin domain containing 3 (NLRP3) Inflammasome: Pathogen-or host-derived signals induce the activation of inflammatory caspases, such as caspase-1/4/5/11. These caspases cleave gasdermin D (GSDMD) resulting in the separation of the N- and C-terminal domains (GSDMD Nterm, GSDMD Cterm). GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death.
2. Apoptosis
Apoptosis is a naturally occurring process that plays a distinct role in morphological and biochemical changes. It is also called PCD and can occur through two distinct apoptotic pathways: the intrinsic and extrinsic pathways [4]. Some of the main characteristics of apoptotic cells include chromatin condensation, DNA fragmentation, phosphatidylserine (PtdSer) cell surface exposure on the outer leaflet of the plasma membrane, and the formation of apoptotic bodies [5]. The key mediators of apoptosis are caspase enzymes [6]. Apoptosis is characterized morphologically by membrane blebbing, chromatin condensation, intra-nucleosomal DNA fragmentation, and the formation of apoptotic bodies and is mediated by a specific cysteine protease family of proteins called caspases [7]. Cytochrome c (Cyt c) is found in the inner membrane areas of healthy mitochondria, where it serves as an electron shuttle in the respiratory chain to interact with cardiolipin (CL) [8][9]. Several proapoptotic stimuli cause the outer membrane to permeabilize, allowing contact between the intermembrane and intercristae gaps and the mobilization of Cyt c from CL, allowing Cyt c release [10]. Cyt c facilitates the allosteric stimulation of apoptosis-protease activating factor-1 (Apaf-1), which is necessary for caspase-9 and caspase-3 proteolytic maturation in the cytosol. Mitochondrial disruption triggers the release of Cyt c, which forms apoptosomes with Apaf-1 and caspase-9 during apoptosis. Dimerization activates caspases in apoptosomes, resulting in caspase signaling. Through the mitochondrion, apoptosis-inducing factor and endonuclease G may also promote caspase-9-independent apoptosis [11].
Caspases that were activated cause apoptotic cells to be dismantled. However, cytosolic Cyt c was linked to important cell processes such as differentiation, implying that its release is not always all-or-nothing and that mitochondrial outer membrane permeabilization does not always result in cell death [9]. Apoptosis is triggered when cell-surface death receptors such as Fas (also known as CD95) are bound by their ligands (the intrinsic pathway) or when pro-apoptotic proteins of the Bcl-2 family cause the permeabilization of the mitochondrial outer membrane (the extrinsic pathway). The activation of both pathways results in the activation of the caspase family of proteases, ultimately resulting in the cell’s destruction [12]. To determine the signaling processes that occur in apoptosis, Chen et al. demonstrated that mitochondrial membrane potential was lost when caspase-9 was dimerized, and anti-apoptotic Bcl-2, Bcl-xL, and Mcl-1 were cleaved. Cleavage-resistant of Bcl-2, Bcl-xL, or Mcl-1 also significantly reduced caspase-9-dependent Cyt c release and mitochondrial membrane potential loss [13].
Natural killer (NK) cells are an immune effector cell population with an intrinsic ability to kill virus-infected and tumor cells without prior antigen sensitization. NK cell-mediated cytotoxicity is regulated by integrating signals from a repertoire of activating and inhibitory receptors. Activated NK cells can kill target cells through different mechanisms, including the release of preformed cytolytic granules and pro-inflammatory cytokines as well as trigging cell apoptosis through death-receptor pathways by inducing death ligands such as the Fas ligand and the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) [14]. In recent investigations, TRAIL has been a useful biomarker for distinguishing between bacterial and viral infections [15]. High levels of interferons (IFNs) were related to lymphocyte death, and TRAIL and its receptor, death receptor 5 (DR5), were suggested as possible molecules [16]. The TRAIL and DR5 expression levels in patient blood samples [17] and IFN alpha/beta expression by plasmacytoid dendritic cells (pDCs) in tonsil tissue are significantly higher in progressors than non-progressors [18].
3. Necroptosis
Necroptosis or programmed necrosis is considered a “trap door” to eliminate pathogens when the caspase system gets impaired [19]. Morphologically, necroptosis is characterized by increased cell volume, membrane rupture and swelling of organelles, cellular collapse, and release of cellular contents [20]. The hallmarks of necrotizing non-apoptotic cell death are regulated necrosis (namely necroptosis) and rapid loss of plasma membrane integrity [21]. Necrosis is regulated by several mediators, including death receptors (DRs) such as tumor necrosis factor receptor-1 (TNFR1), CD95, and DR4/5, IFNs, Toll-like receptors (TLRs), intracellular RNA and DNA sensors. This necrosis is caused by the protein receptor-interacting protein kinase-3 (RIPK3) and mixed lineage kinase domain-like (MLKL) [22]. Recent evidence reveals that necroptosis is associated with RIPK3 and MLKL-induced inflammation [23] and is implicated in the pathogenesis of several diseases such as strokes [24], myocardial infarction [25], and pulmonary disease [26] and host defense against the virus [27]. Activated RIPL3 and MLKL complexes are the key necrosis regulators that orchestrate tissue inflammation and injury. A recent study reported that phosphorylated RIPK3 and MLKL signaling promotes inflammation, airway remodeling, and emphysema in chronic obstructive pulmonary disease (COPD) in the lung tissue of COPD patients [28]. Several other factors are also mainly involved in this mode of cell death. For example, intracellular ATP depletion [29][30], lysosomal enzyme release [31], and excessive ROS generation determine the cell death fate by necrosis [32].
4. Pyroptosis
Pyroptosis is a newly discovered programmed cell death, and it was studied in the context of cancer [33] and neuronal diseases [34]. Morphologically, pyroptosis greatly differs from other cell death mechanisms such as apoptosis and necrosis in terms of occurrence and regulatory mechanisms [35][36][37]. Pyroptosis differs from apoptosis by being highly pro-inflammatory, involving the formation of membrane pores, is lytic and is driven by caspase-1, previously regarded as the “IL-1β-converting enzyme”, which is not associated with apoptosis. Moreover, it differs from other pore-forming RCDs, such as necroptosis, by involving the gasdermin (GSDM) family of proteins as the membrane pore-forming agent [1]. It manifests by activation of one or more caspases, primarily caspase-1 in humans and mice, human caspase-4/5 in humans and caspase-11 in mice [38][39] and thus can broadly be classified into canonical caspase-1 dependent and non-canonical caspase-1 independent pyroptosis. Both the types, however, remain morphologically similar.
4.1. Caspase-1-Dependent Pyroptosis
Caspase-1 occurs as an inactive 45 kDa precursor protein in the cytosol. Multimeric protein complexes within the cytosol, known as inflammasomes, are activated in response to pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs) [40] generated by microbial infection and endogenous stress [41]. Inflammasomes can result in either cytokine processing or pyroptosis. An inflammasome is composed of caspase-1 subfamily caspases such as caspases-1/4/5 in humans and caspases-1/11 in mice. These inflammasome-initiating sensors include NOD-like receptors (NLRP1, NLRP3, NLRC4), IFN-inducible protein AIM2 is also known as absent in melanoma 2 (AIM2) or pyrin, with or without the apoptosis-associated speck-like protein containing a CARD (ASC), an inflammasome adaptor protein [42][43][44]. All three classes of inflammasome sensors and ASC contain CARD (caspase activation and recruitment domains), pyrin domains (PYD), or both. NLRs recognize those danger signals introduced into the host cell cytosol, resulting in a massive release of inflammatory cytokines (IL-1 β, IL-18). Some NLRs, such as NLRC4 [45], can directly bind to caspase-1, while others, including NLRP3, bind to the adapter protein ASC, which contains a CARD domain that interacts and activates with caspase-1, including NLRP3 [46]. Caspase-1 is activated within this complex via dimerization and autoproteolysis [47]. A single NLR [48][49] or multiple NLRs [50] can activate caspase-1. Once activated, caspase-1 also produces plasma-membrane pores of 1.1–2.4 nm in diameter, causing water influx, the release of cellular ionic gradients, cell swelling, and osmotic lysis [51]. The mechanism of the pore formation during pyroptosis, which is still less known, is attributed to the action of a newly discovered component of inflammasomes, gasdermin D (GSDMD) [52]. The pyroptosis execution protein GSDMD is activated by inflammatory caspases (caspase-1/11/4/5), which cleaves GSDMD to expose its N-terminal domain for relocation to the plasma membrane, producing large oligomeric pores [53][54][55][56]. Caspase-1 activates the pro-forms of IL-1β and IL-18 and releases them through the GSDMD pores during pyroptosis, although the exact secretion mechanism remains unclear. The steps that lead to cell death after activation of caspase-1 mediated signaling is not fully understood.
4.2. Caspase-1-Independent Pyroptosis
Caspase-1-independent pyroptosis is considered non-canonical pyroptosis and is activated by apical activators of the inflammasome, including caspases-4/5/11 [57][58][59][60]. Unlike canonical pyroptosis, non-canonical pyroptosis is driven by non-canonical inflammasomes without any cytokine release. However, caspase-1-dependent pyroptosis has reported a high affinity for GSDMD, IL-1β, and IL-18, whereas caspase-1-independent pyroptosis has a comparable affinity for GSDMD, which is less likely to release IL-1β and IL-18 cytokine [52][61]. Activated caspase-11 causes cell swelling, cell membrane damage, free radical production, and inflammation, which are the morphological characteristics of necroptosis. Caspase-11 also turns procaspase-1 into caspase-1 family and caspase-3, which leads to a massive release of inflammatory cytokines such as IL-1β and IL-18 [62]. Additionally, there is exciting evidence that there is a crosstalk between non-canonical and canonical pyroptosis, in which non-canonical inflammasome-mediated activation of GSDMD triggers canonical inflammasome activation and cytokine release [63]. The activation of GSDMD by non-canonical inflammasomes leads to canonical inflammasome activation and cytokine release in the intracellular DAMP generation [64].
References
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541.
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11.
- Hu, X.-M.; Li, Z.-X.; Lin, R.-H.; Shan, J.-Q.; Yu, Q.-W.; Wang, R.-X.; Liao, L.-S.; Yan, W.-T.; Wang, Z.; Shang, L.; et al. Guidelines for Regulated Cell Death Assays: A Systematic Summary, A Categorical Comparison, A Prospective. Front. Cell Dev. Biol. 2021, 9, 634690.
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516.
- Arandjelovic, S.; Ravichandran, K. Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol. 2015, 16, 907–917.
- Saraste, A.; Pulkki, K. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc. Res. 2000, 45, 528–537.
- Thornberry, N.A.; Lazebnik, Y. Caspases: Enemies Within. Science 1998, 281, 1312–1316.
- Basova, L.V.; Kurnikov, I.V.; Wang, L.; Ritov, V.B.; Belikova, N.A.; Vlasova, I.I.; Pacheco, A.A.; Winnica, D.E.; Peterson, J.; Bayir, H.; et al. Cardiolipin Switch in Mitochondria: Shutting off the Reduction of Cytochrome c and Turning on the Peroxidase Activity. Biochemistry 2007, 46, 3423–3434.
- Garrido, C.; Galluzzi, L.; Brunet, M.; Puig, P.E.; Didelot, C.; Kroemer, G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006, 13, 1423–1433.
- Arnoult, D.; Parone, P.; Martinou, J.-C.; Antonsson, B.; Estaquier, J.; Ameisen, J.C. Mitochondrial release of apoptosis-inducing factor occurs downstream of cytochrome c release in response to several proapoptotic stimuli. J. Cell Biol. 2002, 159, 923–929.
- Guerrero, A.D.; Schmitz, I.; Chen, M.; Wang, J. Promotion of Caspase Activation by Caspase-9-mediated Feedback Amplification of Mitochondrial Damage. J. Clin. Cell. Immunol. 2012, 3, 1000126.
- Fulda, S.; Debatin, K.-M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811.
- Chen, M.; Guerrero, A.D.; Huang, L.; Shabier, Z.; Pan, M.; Tan, T.-H.; Wang, J. Caspase-9-induced Mitochondrial Disruption through Cleavage of Anti-apoptotic BCL-2 Family Members. J. Biol. Chem. 2007, 282, 33888–33895.
- Zhu, H.; Blum, R.H.; Bernareggi, D.; Ask, E.H.; Wu, Z.; Hoel, H.J.; Meng, Z.; Wu, C.; Guan, K.-L.; Malmberg, K.-J.; et al. Metabolic Reprograming via Deletion of CISH in Human iPSC-Derived NK Cells Promotes In Vivo Persistence and Enhances Anti-tumor Activity. Cell Stem Cell 2020, 27, 224–237.
- Tsao, Y.-T.; Tsai, Y.-H.; Liao, W.-T.; Shen, C.-J.; Cheng, C.-M. Differential Markers of Bacterial and Viral Infections in Children for Point-of-Care Testing. Trends Mol. Med. 2020, 26, 1118–1132.
- Artykov, A.A.; Yagolovich, A.V.; Dolgikh, D.A.; Kirpichnikov, M.P.; Trushina, D.B.; Gasparian, M.E. Death Receptors DR4 and DR5 Undergo Spontaneous and Ligand-Mediated Endocytosis and Recycling Regardless of the Sensitivity of Cancer Cells to TRAIL. Front. Cell Dev. Biol. 2021, 9, 733688.
- Barblu, L.; Machmach, K.; Gras, C.; Delfraissy, J.-F.; Boufassa, F.; Leal, M.; Ruiz-Mateos, E.; Lambotte, O.; Herbeuval, J.-P. Plasmacytoid Dendritic Cells (pDCs) From HIV Controllers Produce Interferon-α and Differentiate Into Functional Killer pDCs Under HIV Activation. J. Infect. Dis. 2012, 206, 790–801.
- Herbeuval, J.-P.; Nilsson, J.; Boasso, A.; Hardy, A.W.; Kruhlak, M.J.; Anderson, S.A.; Dolan, M.J.; Dy, M.; Andersson, J.; Shearer, G.M. Differential expression of IFN-α and TRAIL/DR5 in lymphoid tissue of progressor versus nonprogressor HIV-1-infected patients. Proc. Natl. Acad. Sci. USA 2006, 103, 7000–7005.
- Kaiser, W.J.; Sridharan, H.; Huang, C.; Mandal, P.; Upton, J.; Gough, P.J.; Sehon, C.A.; Marquis, R.W.; Bertin, J.; Mocarski, E.S. Toll-like Receptor 3-mediated Necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 2013, 288, 31268–31279.
- Belizário, J.; Vieira-Cordeiro, L.; Enns, S. Necroptotic Cell Death Signaling and Execution Pathway: Lessons from Knockout Mice. Mediat. Inflamm. 2015, 2015, 128076.
- Choi, M.E.; Price, D.R.; Ryter, S.W.; Choi, A.M.K. Necroptosis: A crucial pathogenic mediator of human disease. JCI Insight 2019, 4, e128834.
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflamm. 2018, 15, 1–9.
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320.
- Naito, M.G.; Xu, D.; Amin, P.; Lee, J.; Wang, H.; Li, W.; Kelliher, M.; Pasparakis, M.; Yuan, J. Sequential activation of necroptosis and apoptosis cooperates to mediate vascular and neural pathology in stroke. Proc. Natl. Acad. Sci. USA 2020, 117, 4959–4970.
- Luedde, M.; Lutz, M.; Carter, N.; Sosna, J.; Jacoby, C.; Vucur, M.; Gautheron, J.; Roderburg, C.; Borg, N.; Reisinger, F.; et al. RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc. Res. 2014, 103, 206–216.
- Chen, D.; Gregory, A.D.; Li, X.; Wei, J.; Burton, C.L.; Gibson, G.; Scott, S.J.; Croix, C.M.S.; Zhang, Y.; Shapiro, S.D. RIP3-dependent necroptosis contributes to the pathogenesis of chronic obstructive pulmonary disease. JCI Insight 2021, 6, e144689.
- Upton, J.; Kaiser, W.J.; Mocarski, E.S. Virus Inhibition of RIP3-Dependent Necrosis. Cell Host Microbe 2010, 7, 302–313.
- Lu, Z.; Van Eeckhoutte, H.P.; Liu, G.; Nair, P.M.; Jones, B.; Gillis, C.M.; Nalkurthi, B.C.; Verhamme, F.; Buyle-Huybrecht, T.; Vandenabeele, P.; et al. Necroptosis Signaling Promotes Inflammation, Airway Remodeling, and Emphysema in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2021, 204, 667–681.
- Lieberthal, W.; Menza, S.A.; Levine, J.S. Graded ATP depletion can cause necrosis or apoptosis of cultured mouse proximal tubular cells. Am. J. Physiol. Physiol. 1998, 274, F315–F327.
- Eguchi, Y.; Shimizu, S.; Tsujimoto, Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997, 57, 1835–1840.
- Piao, S.; Amaravadi, R.K. Targeting the lysosome in cancer. Ann. N. Y. Acad. Sci. 2016, 1371, 45–54.
- Zhao, J.; Jiang, P.; Guo, S.; Schrodi, S.J.; He, D. Apoptosis, Autophagy, NETosis, Necroptosis, and Pyroptosis Mediated Programmed Cell Death as Targets for Innovative Therapy in Rheumatoid Arthritis. Front. Immunol. 2021, 12, 809806.
- Tan, Y.; Chen, Q.; Li, X.; Zeng, Z.; Xiong, W.; Li, G.; Li, X.; Yang, J.; Xiang, B.; Yi, M. Pyroptosis: A new paradigm of cell death for fighting against cancer. J. Exp. Clin. Cancer Res. 2021, 40, 153.
- Hu, Y.; Wang, B.; Li, S.; Yang, S. Pyroptosis, and its Role in Central Nervous System Disease. J. Mol. Biol. 2022, 434, 167379.
- Zhao, G.; Xie, Z. Pyroptosis and neurological diseases. Neuroimmunol. Neuroinflamm. 2014, 1, 60–65.
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128.
- Fink, S.L.; Cookson, B.T. Apoptosis, Pyroptosis, and Necrosis: Mechanistic Description of Dead and Dying Eukaryotic Cells. Infect. Immun. 2005, 73, 1907–1916.
- Liu, W.; Yang, D.; Shi, J.; Wen, P.; Zhang, J.; Wang, Z.; Hu, B.; Shi, X.; Cao, S.; Guo, W.; et al. Caspase-1 Inhibitor Reduces Pyroptosis Induced by Brain Death in Kidney. Front. Surg. 2021, 8, 760989.
- Man, S.M.; Karki, R.; Briard, B.; Burton, A.; Gingras, S.; Pelletier, S.; Kanneganti, T.-D. Differential roles of caspase-1 and caspase-11 in infection and inflammation. Sci. Rep. 2017, 7, 45126.
- Man, S.M.; Karki, R.; Kanneganti, T.-D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75.
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328.
- McKenzie, B.; Dixit, V.M.; Power, C. Fiery Cell Death: Pyroptosis in the Central Nervous System. Trends Neurosci. 2020, 43, 55–73.
- McKenzie, B.A.; Mamik, M.K.; Saito, L.B.; Boghozian, R.; Monaco, M.C.; Major, E.O.; Lu, J.-Q.; Branton, W.G.; Power, C. Caspase-1 inhibition prevents glial inflammasome activation and pyroptosis in models of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2018, 115, E6065–E6074.
- Rathinam, V.A.; Fitzgerald, K.A. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell 2016, 165, 792–800.
- Poyet, J.-L.; Srinivasula, S.M.; Tnani, M.; Razmara, M.; Fernandes-Alnemri, T.; Alnemri, E.S. Identification of Ipaf, a Human Caspase-1-activating Protein Related to Apaf-1. J. Biol. Chem. 2001, 276, 28309–28313.
- Bergsbaken, T.; Fink, S.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Genet. 2009, 7, 99–109.
- von Moltke, J.; Ayres, J.S.; Kofoed, E.M.; Chavarría-Smith, J.; Vance, R.E. Recognition of Bacteria by Inflammasomes. Annu. Rev. Immunol. 2013, 31, 73–106.
- Faustin, B.; Lartigue, L.; Bruey, J.-M.; Luciano, F.; Sergienko, E.; Bailly-Maitre, B.; Volkmann, N.; Hanein, D.; Rouiller, I.; Reed, J.C. Reconstituted NALP1 Inflammasome Reveals Two-Step Mechanism of Caspase-1 Activation. Mol. Cell 2007, 25, 713–724.
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426.
- Bergsbaken, T.; Fink, S.L.; Hartigh, A.B.D.; Loomis, W.P.; Cookson, B.T. Coordinated Host Responses during Pyroptosis: Caspase-1–Dependent Lysosome Exocytosis and Inflammatory Cytokine Maturation. J. Immunol. 2011, 187, 2748–2754.
- Fink, S.L.; Cookson, B.T. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell. Microbiol. 2006, 8, 1812–1825.
- He, W.-T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298.
- Tsuchiya, K.; Nakajima, S.; Hosojima, S.; Nguyen, D.T.; Hattori, T.; Le, T.M.; Hori, O.; Mahib, M.R.; Yamaguchi, Y.; Miura, M.; et al. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat. Commun. 2019, 10, 2091.
- Ramos-Junior, E.S.; Morandini, A.C. Gasdermin: A new player to the inflammasome game. Biomed. J. 2017, 40, 313–316.
- Sborgi, L.; Rühl, S.; Mulvihill, E.; Pipercevic, J.; Heilig, R.; Stahlberg, H.; Farady, C.J.; Muller, D.J.; Broz, P.; Hiller, S. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016, 35, 1766–1778.
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158.
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192.
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science 2013, 341, 1250–1253.
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszyński, A.; et al. Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science 2013, 341, 1246–1249.
- Gu, Y.; Kuida, K.; Tsutsui, H.; Ku, G.; Hsiao, K.; Fleming, M.A.; Hayashi, N.; Higashino, K.; Okamura, H.; Nakanishi, K.; et al. Activation of Interferon-γ Inducing Factor Mediated by Interleukin-1β Converting Enzyme. Science 1997, 275, 206–209.
- Ding, J.; Shao, F. SnapShot: The Noncanonical Inflammasome. Cell 2017, 168, 544–544.e1.
- Agnew, A.; Nulty, C.; Creagh, E. Regulation, Activation and Function of Caspase-11 during Health and Disease. Int. J. Mol. Sci. 2021, 22, 1506.
- Platnich, J.; Chung, H.; Lau, A.; Sandall, C.F.; Bondzi-Simpson, A.; Chen, T.; Komada, T.; Trotman-Grant, A.C.; Brandelli, J.R.; Chun, J.; et al. Shiga Toxin/Lipopolysaccharide Activates Caspase-4 and Gasdermin D to Trigger Mitochondrial Reactive Oxygen Species Upstream of the NLRP3 Inflammasome. Cell Rep. 2018, 25, 1525–1536.
- Downs, K.P.; Nguyen, H.; Dorfleutner, A.; Stehlik, C. An overview of the non-canonical inflammasome. Mol. Asp. Med. 2020, 76, 100924.
More
Information
Subjects:
Immunology; Pathology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.3K
Revisions:
2 times
(View History)
Update Date:
05 Jul 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No