 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | César Augusto Correia De Sequeira | -- | 3438 | 2022-07-01 12:32:34 | | | |
| 2 | Jason Zhu | + 1 word(s) | 3439 | 2022-07-04 05:56:47 | | | | |
| 3 | Jason Zhu | -1 word(s) | 3438 | 2022-07-07 04:19:24 | | | | |
| 4 | Jason Zhu | Meta information modification | 3438 | 2022-07-08 03:49:35 | | |
Video Upload Options
Leaching is a central unit operation in the hydrometallurgical processing of minerals, which often occurs by means of electrochemical reactions. Application of mixed potential theory to explain the kinetics of oxidative and reductive leaching processes is a useful concept in explaining observed results. Native metals, selected oxides, and most base metal sulfides are electron-conducting phases. For these minerals, leaching may take place by normal corrosion, passivation or galvanic couple mechanisms, which provide individual electrode kinetics enabling the calculation of mixed potentials and overall reaction kinetics.
1. Leaching of Galena
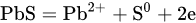
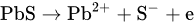
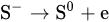
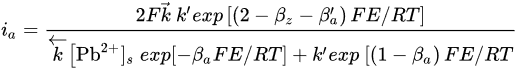 where [Pb2+]S is the concentration of lead ions at the electrode surface, k⃗ and k are the respective forward and reverse rate constants for Reaction, k′ is the specific rate of Reaction and the other terms are analogous to those previously defined. At sufficiently positive values of E or at low [Pb2+], the expression for ia reduces to
where [Pb2+]S is the concentration of lead ions at the electrode surface, k⃗ and k are the respective forward and reverse rate constants for Reaction, k′ is the specific rate of Reaction and the other terms are analogous to those previously defined. At sufficiently positive values of E or at low [Pb2+], the expression for ia reduces to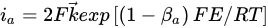
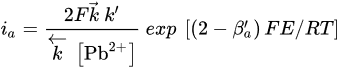

2. Leaching of Gold and Silver
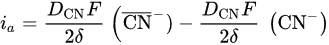
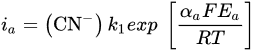
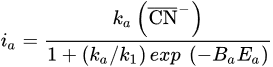
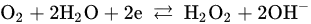
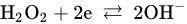

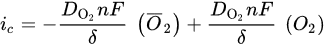
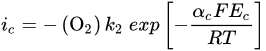

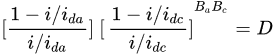
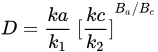
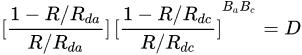
3. Leaching of Pyrite
Pyrite, the most common sulfide mineral, dissolves by the following reactions:

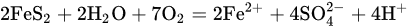
The overall reaction can be written in terms of the half-reactions for the oxidation of pyrite and the reduction of ferric ions and oxygen. Holmer and Crundwell [18] investigated each of these half-reactions and investigated the overall reaction. These measurements showed that the kinetics of the half-reaction for the anodic dissolution of pyrite is given by:
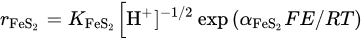
and that the half-reaction for the reduction of ferric ions is given by:

Here E is the mixed potential, after the corrosion theory of Wagner and Traud (1938) [19]. The order of the reaction of—0.5 with respect to H+ for the anodic reaction indicates that reaction consists of some steps, possibly involving adsorbed hydroxide ions, prior to the rate-determining step. Considering that every electron donated by the anodic dissolution of pyrite is instantaneously accepted by the reduction of ferric ions, then τFeS2=τFe and

The substitution of this equation for the mixed potential into Equation yields the rate equation for the oxidative dissolution of pyrite by ferric ions. When [Fe2+] is greater than 0.001 M, i.e., KFeS2[H+]−1/2 is much less than K′Fe[Fe2+], then this equation is given by
This equation predicts that the rate of dissolution of pyrite is one-half order in ferric ions and negative one-half order in H+. Results of recent investigations of the rate of dissolution of pyrite by ferric ions are correctly described by the electrochemical mechanism derived by Holmes and Crundwell [18]. They also measured the cathodic reduction of oxygen on pyrite, and showed that the order of reaction with respect to H+ is 0.14. Using this result, they derived an expression for the rate of dissolution of pyrite in the presence of oxygen, which is given as follows:

This result is again related to the electrochemical mechanism of Holmes and Crundwell [18], and was confirmed by Rimstidt and Vaughan [20]. The effect of the concentrations of ferric and ferrous ions on the mixed potential of pyrite showed that the slope of the mixed potential is 0.059 mV/decade, which is in good agreement with the theoretical value. If KFeS2[H+]−1/2 is much less than K′Fe[Fe2+], then the predicted value of the slope of the mixed potential is—0.059 mV/decade, which is close to the experimentally determined value of—0.056 mV/decade. Thus, once again, the experiments of the mixed potential confirm the theory.
4. Leaching of Sphalerite
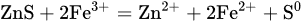
whose rate of reaction can be described by the following kinetic expression: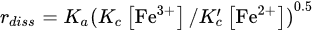
Crundwell [21], using principles of quantum electrochemistry, derived the following expressions for the rates of the anodic half-reaction and the cathodic half-reaction
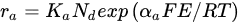
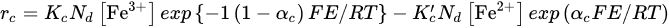
where Nd is the concentration of iron in the sphalerite (mol Fe/mol Zn). Eliminating E between Equations, it can be obtained the rate of the surface reaction:

5. Leaching of Chalcopyrite

-
The number of electrons (seven) transferred per mole copper during anodic oxidation is similar for the ammonia-ammonium sulphate and ammonia-ammonium carbonate solutions;
-
Ammonia-ammonium perchlorate solutions promote a five-electron transfer/copper reaction, possibly forming elemental sulfur on the mineral surface;
-
Ammonium sulphate leaching results in the formation of a Fe-oxyhydroxide layer with low sulfur on the mineral surface;
-
Ammonia-ammonium carbonate solutions resulted in marginal accumulation of iron on the mineral surface, but no formation of a layer was observed;
-
Ammonium perchlorate leaching results in the formation of a Fe-oxyhydroxide layer with moderate sulfur on the mineral surface;
-
The surface product was largely amorphous (90%) and significantly more porous (9–12 times) than unleached chalcopyrite. The observed morphology of the surface product suggests that it is formed through secondary precipitation rather than as part of the chalcopyrite dissolution mechanism;
-
Surface abrasion allows for the removal of the surface product, leading to improved leaching recoveries;
-
The abraded surface product from the small particles leaching experiment contained no sulfur, while surface products found on the stationary block of mineral contained small quantities of sulfur.
References
- Eadington, P.; Prosser, A. Oxidation of lead sulfide in aqueous suspension. IMM Trans. Sect. C 1969, 78, 74–82.
- Pridmore, D.F.; Shuey, R.T. The electrical conductivity of galena, pyrite and chalcopyrite. Am. Mineral. 1976, 61, 248–259.
- Gurin, G.; Titov, K.; Ilyin, Y.; Tarasov, A. Induced polarization of disseminated electronically conductive minerals: A semi-empirical model. Geophys. J. Int. 2015, 200, 1555–1565.
- Dusabermariya, C.; Qian, W.; Bagaragaga, R.; Faruwa, A.; Ali, M. Some experiences of resistivity and induced polarization methods on the exploration of sulfide: A review. J. Geosci. Environ. Prot. 2020, 8, 68–92.
- Paul, R.L.; Nicol, M.J.; Diggle, J.W.; Saunders, A.P. The electrochemical behaviour of galena (lead sulphide)-1. Anodic dissolution. Electrochim. Acta 1978, 23, 625–633.
- Baba, A.A.; Adekola, F. Comparative analysis of the dissolution kinetics of galena in binary solutions of HCl/FeCl3 and HCl/H2O2. Int. J. Miner. Metall. Mater. 2011, 18, 1–9.
- Dutrizac, J.E. The dissolution of galena in ferric chloride media. Metall. Trans. B 1986, 17, 5–17.
- Fuerstenau, M.C.; Chen, C.C.; Han, K.M.; Palmer, B.R. Kinetics of galena dissolution in ferric chloride solutions. Metall. Trans. B 1986, 17, 415–423.
- Chen, A.A. Kinetics of Leaching Galena Concentrates with Ferric Fluosilicate Solution. Master Eng. Thesis, University of British Columbia, BC, Canada, 1992.
- Azizi, A. Gold cyanidation revisited-Kinetic & electrochemical studies of gold-sulfidic ore mixed/multilayer fixed beds. Ph.D. Thesis, University Laval, Quebec City, QC, Canada, 2011.
- Wilkominsky, I.; Rojas, N.; Balladares, E. Gold and silver cyanidation from a residue produced by leaching dead-roasted copper cohite metal. Can. Metall. Q. 2010, 49, 29–37.
- Medina, D.; Anderson, C.G. A review of cyanidation treatment of copper-gold ores and concentrates. Materials 2020, 10, 897.
- Azizi, A.; Ghardrahmati, R. Optimizing and evaluating the operational factors affecting the cyanide leaching circuit of the Aghdareh gold processing plant using a CCD model. Proc. R. Soc. A 2015, 471, 20150681.
- Birich, A.; Stopic, S.; Friedrich, B. Kinetic investigation and dissolution behaviour of cyanide alternative gold leaching. Sci. Rep. 2019, 9, 7191.
- Sabir, S. Silver Hydrometallurgy: Recovery and Recycling; Nova Publishers: Riyadh, Saudi Arabia, 2017.
- Crundwell, F.K. The dissolution and leaching of minerals. Mechanisms, myths and misunderstandings. Hydrometallurgy 2013, 139, 132–148.
- Kudryk, V.; Kellog, H.H. Mechanism and rate-controlling factors in the dissolution of gold in cyanide solution. JOM 1954, 6, 541–548.
- Holmes, P.R.; Crundwell, E.K. The kinetics of the oxidation of pyrite by ferric ions and dissolved oxygen: An electrochemical study. Geochim. Cosmochim. Acta 2000, 64, 263–274.
- Sequeira, C.A.C. High Temperature Corrosion: Fundamentals and Engineering; John Wiley & Sons: Hoboken, NJ, USA, 2019.
- Rimstidt, J.D.; Vaughan, D.J. Pyrite oxidation: A state-of-the-art assessment of the reaction mechanism. Geochim. Cosmochim. Acta 2003, 67, 873–880.
- Crundwell, F.K. Effect of iron impurity in zinc sulfide concentrate on the rate of dissolution. AICHE J. 1988, 34, 1128–1134.
- Lu, Z.Y.; Jeffrey, M.I.; Lawson, F. An electrochemical study of the effect of chloride ions on the dissolution of chalcopyrite in acidic solutions. Hydrometallurgy 2000, 56, 145–155.
- Hua, X.; Zheng, Y.; Xu, Q.; Lu, X.; Cheng, H.; Zou, X.; Song, Q.; Ning, Z.; Free, M.L. Leaching mechanism and electrochemical oxidation on the surface of chalcopyrite in ammonia-ammonium chloride solution. J. Electrochem. Soc. 2018, 165, E466–E476.
- Tanne, C.K.; Schippers, A. Electrochemical investigation of chalcopyrite (bio) leaching residues. Hydrometallurgy 2019, 187, 8–17.
- Moyo, T. An Electrochemical and Leach Study of the Oxidative Dissolution of Chalcopyrite in Ammoniacal Solutions. Ph.D. Thesis, University of Cape Town, Rondebosch, Cape Town, South Africa, 2016.
- Eghbalnia, M. Electrochemical and Raman Investigation of Pyrite and Chalcopyrite Oxidation. Ph.D. Thesis, University of British Columbia, Vancouver, Canada, 2012.
- Asgari, K.; Hassanzadeh, A.; Nazari, S.; Kakylabad, A.B.; Hosseinzadeh, M. Effect of externally adding pyrite and electrical current on galvanic leaching of chalcopyrite concentrate. Physicochem. Probl. Miner. Process. 2021, 57, 106–120.
- Liu, Q.; Chen, M.; Zheng, K.; Yang, Y.; Feng, X.; Li, H. In situ electrochemical investigation of pyrite assisted leaching of chalcopyrite. J. Electrochem. Soc. 2018, 165, H813–H819.
- Solis Marcial, O.J.; Nájera Bastida, A.; Banuelos, J.E.; Valdés Martinez, O.U.; Luevano, L.A.; Serrano Rosales, B. Chacopyrite leaching kinetics in the presence of methanol. Int. J. Chem. Reactor Eng. 2019, 17, 20190081.
- Arena, F.A.; Suegama, P.H.; Bevilaqua, D.; dos Santos, A.L.A.; Fugivara, C.S.; Benedetti, A.V. Simulating the main stages of chalcopyrite leaching and bioleaching in ferrous ions solution: An electrochemical impedance study with a modified carbon paste electrode. Miner. Eng. 2016, 92, 229–241.
- Peng, T.; Liao, W.; Wang, J.; Miao, J.; Peng, Y.; Gu, G.; Wu, X.; Qiu, G.; Zeng, W. Bioleaching and electrochemical behavior of chalcopyrite by a mixed culture at low temperature. Front. Microbiol. 2021, 12, 663757.
- Sequeira, C.A.C.; Santos, D.M.F.; Chen, Y.; Anastassakis, G. Chemical metathesis of chalcopyrite in acidic solutions. Hydrometallurgy 2008, 92, 135–140.
- Sequeira, C.A.C.; Santos, D.M.F. Transient film formation on chalcopyrite in acidic solutions. J. Appl. Electrochem. 2010, 40, 123–131.
- Asselin, E. Thermochemistry of the Fe, Ni and Co-NH3-H2O system as they relate to the Caron process: A review. Min. Metall. Process. 2011, 28, 169–175.
- Sequeira, C.A.C. Electrohydrometallurgical Recovery of Cadmium and Nickel from Spent Batteries. In Mineral Processing and the Environment; Gallios, G.P., Matis, K.A., Eds.; NATO ASI Series 2: Environment Kluwer; Academic Publishers: Dordrecht, The Netherlands, 1998; Volume 43, pp. 129–142.
- Brito, P.S.D.; Patricio, S.; Rodrigues, L.F.; Santos, D.M.F.; Sequeira, C.A.C. Electrodeposition of Zn-Mn alloys from recycling battery leach solutions in the presence of amines. In The Sustainable World-WIT Transactions on Ecology and the Environment; Brebbia, C.A., Ed.; WIT Press, Wessex, Institute of Technology: Southampton, UK, 2010; Volume 142, pp. 367–378.
- Sousa, N.R.; Borges, P.M.R.; Magueijo, V.M.; Brito, P.S.D.; Sequeira, C.A.C. Electrolytic reactors for the recovery of cadmium from leaching solutions. Key Eng. Mater. 2002, 230–232, 416–419.
- Rademan, J.A.M.; Lorenzen, L.; Van Deventer, J.S.J. The leaching characteristics of Ni-Cu matte in the acid-oxygen pressure leach process at Impala platinum. Hydrometallurgy 1999, 52, 231–252.
- Nikkhou, F.; Xia, F.; Yao, X.; Adegoks, I.A.; Gu, Q.; Kimpton, J.A. A flow-through reaction cell for studying minerals leaching by in-situ synchrotron powder X-ray diffraction. Minerals 2020, 10, 990.
- Sander, M.; Hofstetter, T.B.; Gorski, C.A. Electrochemical analyses of redox-active iron minerals: A review of nonmediated and mediated approaches. Environ. Sci. Technol. 2015, 49, 5862–5878.
- Gow, R.N.V. Spectroelectrochemistry and Modeling of Enargite (Cu3AsS4) Reactivity under Atmospheric Conditions. Ph.D. Thesis, University of Montana, Missoula, Butte, MT, USA, 2015.
- Yessengaziyev, A.; Kenzhaliyev, B.; Berkinbayeva, A.; Sharipov, R.; Suleimenov, E. Electrochemical extraction of Pb and Zn from a collective concentration using a sulfur-grafite electrode as a cathode. J. Chem. Technol. Metall. 2017, 52, 975–980.
- Pugaev, D.; Nicol, M.; Senanayake, G. The mechanisms of the passivation of sulfide minerals in oxidative leaching processes. In Proceedings of the 6th Southern African Base Metals Conference, Phalaborwa, South Africa, 18–20 July 2011; pp. 39–48.
- Moreno-Saldaña, S.I.; Martinez-Gómez, V.J.; Valle-Cervantes, S.; Lucho-Chigo, R.; Rojas-Montes, J.C.; Fuentes-Aceituno, J.C.; Pérez-Garibay, R. Analysis of galena leaching and maximum electrodeposition capacity of Pb using an electrochemical cell. JOM 2021, 73, 1353–1361.
- Chaerun, S.K.; Putri, E.A.; Mubarok, M.Z. Bioleaching of indonesian galena concentrate with an iron- and sulfur-oxidizing mixotrophic bacterium at room temperature. Front. Microbiol 2020, 11, 557548.
- Zhang, Z.; Liu, B.; Wu, M.; Sun, L. An electrochemical method to investigate the effects of compound composition on gold dissolution in thiosulfate solution. Green Proc. Synth. 2020, 9, 496–502.
- Sun, C.B.; Zhang, X.L.; Kou, J.; Xing, Y. A review of gold extraction using noncyanide lixiviants: Fundamentals, advancements, and challenges toward alkaline-sulfur containing leaching agents. Int. J. Miner. Metall. Mater. 2020, 27, 417–431.
- Sanchez-Ortiz, W.; Aldana-González, J.; Monh, T.L.; Romero-Romo, M.; Mejia-Caballero, I.; Ramirez-Silva, T.; Arce-Estrada, E.M.; Mugica Álvarez, V.; Palomar-Pardavé, M. A deep eutectic solvent as leaching agent and electrolytic bath for silver recovery from spent silver oxide batteries. J. Electrochem. Soc. 2021, 168, 016508.
- Reyes-Sandoval, E.; Fuentes-Aceituno, J.C. A study of the metallic silver dissolution with the MEA-NH3-Cu system. Rev. Matéria 2018, 23, e-12004.
- Tanne, C.; Schippers, A. Electrochemical investigation of microbially and galvanically leached chalcopyrite. Hydrometallurgy 2021, 202, 105603.
- Ahmed, M.; Hussein, I.A.; Onawole, A.T.; Saad, M.-A.; Khaled, M. Electrochemical removal of pyrite scale using green formulations. Sci. Rep. 2021, 11, 4796.
- Ma, Y.; Yang, Y.; Gao, X.; Fan, R.; Chen, M. The galvanic effect of pyrite enhanced (bio)leaching of enargite, Cu3 As S4. Hydrometallurgy 2021, 202, 105613.
- Lundstrom, M. Chalcopyrite Dissolution in Cupric Chloride Solutions; Helsinki University of Technology: Helsinki, Finland, 2009.
- Dizer, O.; Rogozhnikov, D.; Karimov, K.; Kuzas, E.; Suntsov, A. Nitric acid dissolution of tenantite, chalcopyrite and sphalerite in the presence of Fe (III) ions and FeS2. Materials 2022, 15, 1545.
- Tafoya-Medina, N.A.; Chuck-Hernandez, C.; Medina, D.I. Study of the electrooxidation of a zinc concentrate. Materials 2021, 14, 2868.
- Alonso, A.R.; Lapidus, G.T.; González, I. A strategy to determine the potential interval for selective silver electrodeposition from ammoniacal thiosulfate solutions. Hydrometallurgy 2007, 85, 144–153.


