 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hung yu Huang | -- | 2043 | 2022-06-23 12:51:55 | | | |
| 2 | Lindsay Dong | + 3 word(s) | 2046 | 2022-06-24 02:53:24 | | |
Video Upload Options
Prolonged mechanical ventilation (PMV) is defined as successful extubation after more than three spontaneous breathing trials or taking more than 14 days. The pathophysiology of PMV includes the presence of an abnormal respiratory drive or ventilator-induced diaphragm dysfunction. Numerous studies have demonstrated that ventilator-induced diaphragm dysfunction is related to increases in in-hospital deaths, nosocomial pneumonia, oxidative stress, lung tissue hypoxia, ventilator dependence, and costs.
1. Introduction
A percentage of critically ill patients experience chronic respiratory failure and require prolonged mechanical ventilation (PMV) [1]. PMV is defined as successful extubation after more than three spontaneous breathing trials or taking more than 14 days [1], although some studies have used different definitions, especially studies in the United Kingdom and Europe [2]. Approximately 5–13% of patients with acute respiratory failure require PMV, and a trend of increasing PMV exists worldwide [3].
Patients receiving PMV may experience complications, including limb muscle atrophy, impaired functional status, and diaphragm dysfunction [4]. Moreover, the basal respiratory drive levels may be altered in patients with PMV, depending on the pulmonary disease present and the cause of respiratory failure [5][6][7]. Brain stem lesions may impair the central and low respiratory drives, causing abnormal respiratory function, hypoventilation, or respiratory acidosis, any of which may lead to ventilator dependence [7].
2. Respiratory Drive
3. Ventilator-Induced Diaphragm Dysfunction
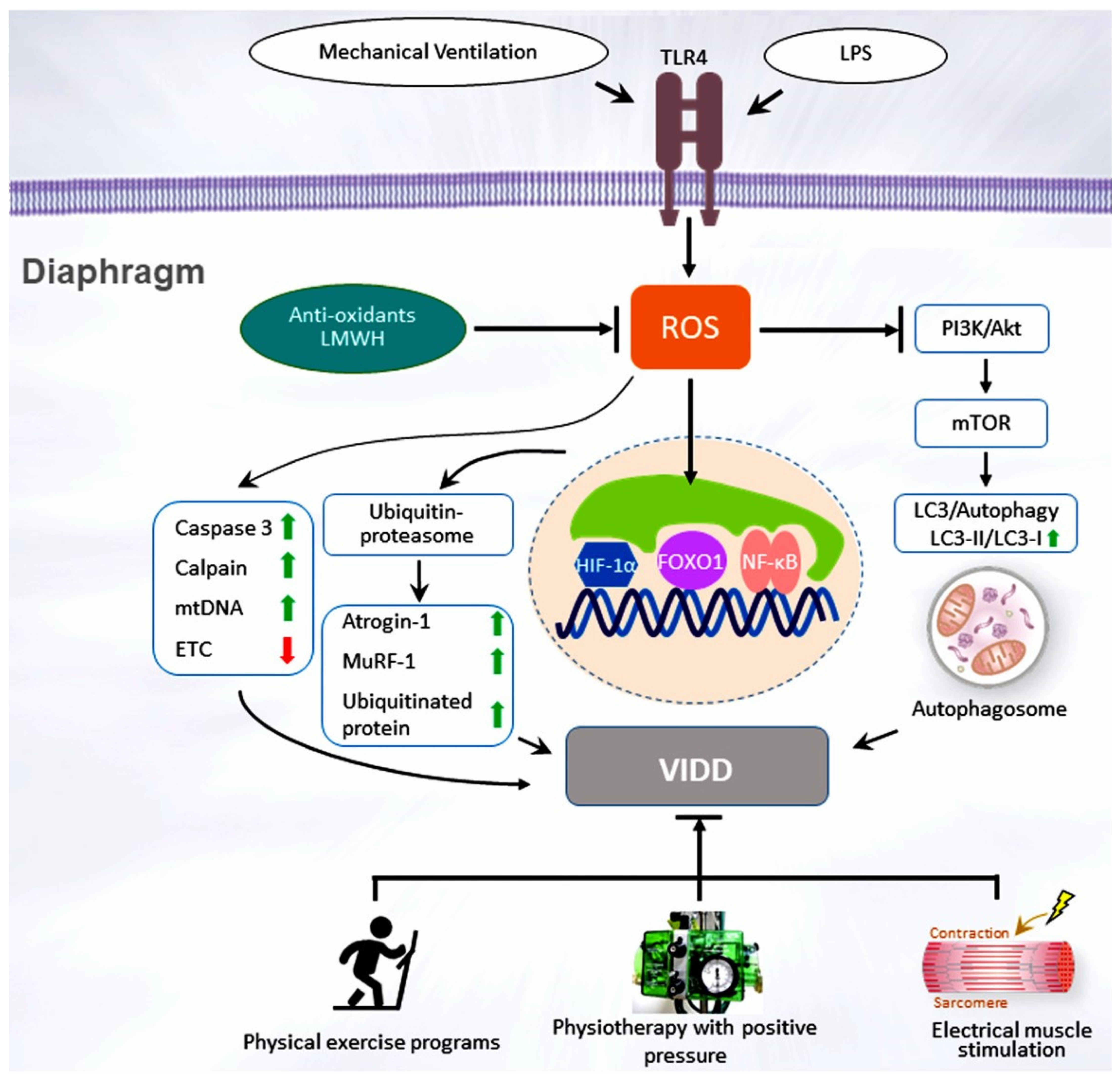
4. Clinical Impact and Multiple Comorbidities
50% of patients (95% confidence interval [CI]: 47–53) were successfully weaned from MV, in-hospital mortality was 29% (95% CI: 26–32), and only 19% (95% CI: 16–24) of patients were discharged home [1]. Recognized factors of weaning failure are underlying respiratory diseases, previous ICU admissions, a high acute physiology and chronic health evaluation (APACHE II) score, and pneumonia [49][50][51][52]. Other factors related to unsuccessful weaning are elevated blood urea nitrogen levels, low Glasgow coma scale (GCS) scores, low serum albumin, and low maximal inspiratory pressure levels [53].
The prognosis for patients requiring PMV is poor. Among 29 studies of PMV, the pooled mortality rate was 62% at year 1 [1]. In a cohort study in the United States, 53.7% of patients requiring PMV were successfully weaned from ventilation at discharge, and 66.9% of these patients were still alive at year 1 [54]. However, the survival rate of patients with ventilator dependence was only 16.4% at post-discharge year 1 [54].
Comorbidities exacerbate diseases and have a severe impact on the outcomes of patients requiring PMV. Chronic obstructive pulmonary disease (COPD), cardiac disease, cerebral vascular or neuromuscular disease, end-stage renal disease, and malignancy are common comorbidities in patients requiring PMV [53][55][56][57][58].
Patients requiring PMV often experience long-term immobilization complications, including limb muscle atrophy, diaphragm dysfunction, pressure ulcers, joint contracture, and deconditioning [59][60]. Muscle-strengthening activities aim to progressively improve mobility and functional activity [61]. Pulmonary rehabilitation has been used to improve physical capacity and the quality of life in patients with chronic pulmonary diseases [62][63][64][65]. For patients requiring PMV, pulmonary rehabilitation could provide clinical benefits [61][66][67][68][69].
A comprehensive physical exercise program includes cardiopulmonary endurance exercises and peripheral muscle training. Upper and lower limb exercises include passive leg raising, weighted resistance, and stationary cycle ergometry training [61][70][71]. Respiratory muscle training consists of placing a sandbag on the abdomen during breathing, using a threshold device, or performing diaphragmatic breathing control [61][69][70][71][72][73][74]. The goal is for the patient to increase their mobility and functional activity and gradually recover from being bedridden to sitting, standing, and walking [67][70][73][74]. The benefits of physical exercise programs for patients requiring PMV include functional improvement, increased weaning rates, shorter duration of hospitalization, and reduced mortality [67][70][73][74]. Notably, Dong et al. showed that early rehabilitation training lessened the diaphragm dysfunction during ventilator use, increased weaning rates from the ventilator, and shortened the intubation duration in patients with MV in the ICU [70].
In patients requiring PMV, immobility and long-term ventilator use result in complications, such as the atrophy of respiratory muscles, decreased lung volume, and atelectasis [4]. When performing rehabilitation exercises, patients requiring PMV may experience respiratory muscle fatigue and intolerance to exercise because of the increased ventilatory demand [66]. Intermittent positive pressure breathing (IPPB) can increase the delivery of inspiratory positive pressure to the airway to achieve homogeneous gas distribution, recruit collapsed alveoli, and facilitate lung expansion [75]. During the weaning process, IPPB and positive end-expiratory pressure (PEEP) may increase lung volume and reduce the work of breathing during the expiratory phase, thus helping patients requiring PMV to tolerate the rehabilitation program [76].
References
- Damuth, E.; Mitchell, J.A.; Bartock, J.L.; Roberts, B.W.; Trzeciak, S. Long-term survival of critically ill patients treated with prolonged mechanical ventilation: A systematic review and meta-analysis. Lancet Respir. Med. 2015, 3, 544–553.
- Rose, L.; McGinlay, M.; Amin, R.; Burns, K.E.; Connolly, B.; Hart, N.; Jouvet, P.; Katz, S.; Leasa, D.; Mawdsley, C.; et al. Variation in Definition of Prolonged Mechanical Ventilation. Respir. Care 2017, 62, 1324–1332.
- Zilberberg, M.D.; de Wit, M.; Pirone, J.R.; Shorr, A.F. Growth in adult prolonged acute mechanical ventilation: Implications for healthcare delivery. Crit. Care Med. 2008, 36, 1451–1455.
- Goligher, E.C.; Dres, M.; Fan, E.; Rubenfeld, G.D.; Scales, D.C.; Herridge, M.S.; Vorona, S.; Sklar, M.C.; Rittayamai, N.; Lanys, A.; et al. Mechanical Ventilation–induced Diaphragm Atrophy Strongly Impacts Clinical Outcomes. Am. J. Respir. Crit. Care Med. 2018, 197, 204–213.
- Hilbert, G.; Gruson, D.; Portel, L.; Vargas, F.; Gbikpi-Benissan, G.; Cardinaud, J.P. Airway occlusion pressure at 0.1 s (P0.1) after extubation: An early indicator of postextubation hypercapnic respiratory insufficiency. Intensive Care Med. 1998, 24, 1277–1282.
- Sato, R.; Hasegawa, D.; Hamahata, N.T.; Narala, S.; Nishida, K.; Takahashi, K.; Sempokuya, T.; Daoud, E.G. The predictive value of airway occlusion pressure at 100 msec (P0.1) on successful weaning from mechanical ventilation: A systematic review and meta-analysis. J. Crit. Care 2021, 63, 124–132.
- Wu, Y.-K.; Lee, C.-H.; Shia, B.-C.; Tsai, Y.-H.; Tsao, T.C.Y. Response to hypercapnic challenge is associated with successful weaning from prolonged mechanical ventilation due to brain stem lesions. Intensive Care Med. 2008, 35, 108–114.
- Benarroch, E.E. Brainstem respiratory chemosensitivity: New insights and clinical implications. Neurology 2007, 68, 2140–2143.
- Montgomery, A.B.; Holle, R.H.; Neagley, S.R.; Pierson, D.J.; Schoene, R.B. Prediction of successful ventilator weaning using airway occlusion pressure and hypercapnic challenge. Chest 1987, 91, 496–499.
- De Jonghe, B.; Bastuji-Garin, S.; Durand, M.C.; Malissin, I.; Rodrigues, P.; Cerf, C.; Outin, H.; Sharshar, T.; Groupe de Reflexion et d’Etude des Neuromyopathies en Reanimation. Respiratory weakness is associated with limb weakness and delayed weaning in critical illness. Crit. Care Med. 2007, 35, 2007–2015.
- Raurich, J.M.; Rialp, G.; Ibáñez, J.; Campillo, C.; Ayestarán, I.; Blanco, C. Hypercapnia test as a predictor of success in spontaneous breathing trials and extubation. Respir. Care 2008, 53, 1012–1018.
- Lee, C.-S.; Chen, N.-H.; Chuang, L.-P.; Chang, C.-H.; Li, L.-F.; Lin, S.-W.; Huang, H.-Y. Hypercapnic Ventilatory Response in the Weaning of Patients with Prolonged Mechanical Ventilation. Can. Respir. J. 2017, 2017, 7381424.
- Gattinoni, L.; Tonetti, T.; Cressoni, M.; Cadringher, P.; Herrmann, P.; Moerer, O.; Protti, A.; Gotti, M.; Chiurazzi, C.; Carlesso, E.; et al. Ventilator-related causes of lung injury: The mechanical power. Intensive Care Med. 2016, 42, 1567–1575.
- Chiumello, D.; Gotti, M.; Guanziroli, M.; Formenti, P.; Umbrello, M.; Pasticci, I.; Mistraletti, G.; Busana, M. Bedside calculation of mechanical power during volume- and pressure-controlled mechanical ventilation. Crit. Care 2020, 24, 417.
- Parhar, K.K.S.; Zjadewicz, K.; Soo, A.; Sutton, A.; Zjadewicz, M.; Doig, L.; Lam, C.; Ferland, A.; Niven, D.J.; Fiest, K.M.; et al. Epidemiology, Mechanical Power, and 3-Year Outcomes in Acute Respiratory Distress Syndrome Patients Using Standardized Screening. An Observational Cohort Study. Ann. Am. Thorac. Soc. 2019, 16, 1263–1272.
- Paudel, R.; Trinkle, C.A.; Waters, C.M.; Robinson, L.E.; Cassity, E.; Sturgill, J.L.; Broaddus, R.; Morris, P.E. Mechanical Power: A New Concept in Mechanical Ventilation. Am. J. Med. Sci. 2021, 362, 537–545.
- O’Leary, A.J.; Drummond, S.E.; Edge, D.; O’Halloran, K.D. Diaphragm Muscle Weakness Following Acute Sustained Hypoxic Stress in the Mouse Is Prevented by Pretreatment with N-Acetyl Cysteine. Oxidative Med. Cell. Longev. 2018, 2018, 4805493.
- Jung, B.; Moury, P.H.; Mahul, M.; De Jong, A.; Galia, F.; Prades, A.; Albaladejo, P.; Chanques, G.; Molinari, N.; Jaber, S. Diaphragmatic dysfunction in patients with ICU-acquired weakness and its impact on extubation failure. Intensive Care Med. 2016, 42, 853–861.
- Dot, I.; Pérez-Teran, P.; Samper, M.-A.; Masclans, J.-R. Diaphragm Dysfunction in Mechanically Ventilated Patients. Arch. Bronconeumol. 2017, 53, 150–156.
- Dres, M.; Goligher, E.; Heunks, L.M.A.; Brochard, L.J. Critical illness-associated diaphragm weakness. Intensive Care Med. 2017, 43, 1441–1452.
- Vassilakopoulos, T. Ventilator-induced diaphragm dysfunction: The clinical relevance of animal models. Intensive Care Med. 2007, 34, 7–16.
- Li, L.-F.; Yu, C.-C.; Huang, H.-Y.; Wu, H.-P.; Chu, C.-M.; Huang, C.-Y.; Liu, P.-C.; Liu, Y.-Y. Suppression of Hypoxia-Inducible Factor 1α by Low-Molecular-Weight Heparin Mitigates Ventilation-Induced Diaphragm Dysfunction in a Murine Endotoxemia Model. Int. J. Mol. Sci. 2021, 22, 1702.
- Peñuelas, O.; Keough, E.; López-Rodríguez, L.; Carriedo, D.; Gonçalves, G.; Barreiro, E.; Lorente, J. Ángel Ventilator-induced diaphragm dysfunction: Translational mechanisms lead to therapeutical alternatives in the critically ill. Intensive Care Med. Exp. 2019, 7, 48.
- Demoule, A.; Molinari, N.; Jung, B.; Prodanovic, H.; Chanques, G.; Matecki, S.; Mayaux, J.; Similowski, T.; Jaber, S. Patterns of diaphragm function in critically ill patients receiving prolonged mechanical ventilation: A prospective longitudinal study. Ann. Intensive Care 2016, 6, 75.
- Smuder, A.J.; Sollanek, K.J.; Nelson, W.B.; Min, K.; Talbert, E.E.; Kavazis, A.N.; Hudson, M.B.; Sandri, M.; Szeto, H.H.; Powers, S.K. Crosstalk between autophagy and oxidative stress regulates proteolysis in the diaphragm during mechanical ventilation. Free Radic. Biol. Med. 2018, 115, 179–190.
- Saccheri, C.; Morawiec, E.; Delemazure, J.; Mayaux, J.; Dubé, B.-P.; Similowski, T.; Demoule, A.; Dres, M. ICU-acquired weakness, diaphragm dysfunction and long-term outcomes of critically ill patients. Ann. Intensive Care 2020, 10, 1.
- Moroz, N.; Maes, K.; Leduc-Gaudet, J.-P.; Goldberg, P.; Petrof, B.J.; Mayaki, D.; Vassilakopoulos, T.; Rassier, D.; Gayan-Ramirez, G.; Hussain, S.N. Oxidants Regulated Diaphragm Proteolysis during Mechanical Ventilation in Rats. Anesthesiology 2019, 131, 605–618.
- Duan, H.; Bai, H. Is Mitochondrial Oxidative Stress the Key Contributor to Diaphragm Atrophy and Dysfunction in Critically Ill Patients? Crit. Care Res. Pract. 2020, 2020, 8672939.
- Hussain, S.N.A.; Mofarrahi, M.; Sigala, I.; Kim, H.C.; Vassilakopoulos, T.; Maltais, F.; Bellenis, I.; Chaturvedi, R.; Gottfried, S.B.; Metrakos, P.; et al. Mechanical Ventilation–induced Diaphragm Disuse in Humans Triggers Autophagy. Am. J. Respir. Crit. Care Med. 2010, 182, 1377–1386.
- Horn, A.G.; Davis, R.T., 3rd; Baumfalk, D.R.; Kunkel, O.N.; Bruells, C.S.; McCullough, D.J.; Opoku-Acheampong, A.B.; Poole, D.C.; Behnke, B.J. Impaired diaphragm resistance vessel vasodilation with prolonged mechanical ventilation. J. Appl. Physiol. 2019, 127, 423–431.
- Smuder, A.J.; Sollanek, K.J.; Min, K.; Nelson, W.B.; Powers, S.K. Inhibition of forkhead boxO-specific transcription prevents mechanical ventilation-induced diaphragm dysfunction. Crit. Care Med. 2015, 43, e133–e142.
- Hyatt, H.W.; Ozdemir, M.; Yoshihara, T.; Nguyen, B.L.; Deminice, R.; Powers, S.K. Calpains play an essential role in mechanical ventilation-induced diaphragmatic weakness and mitochondrial dysfunction. Redox Biol. 2021, 38, 101802.
- Smuder, A.J.; Hudson, M.B.; Nelson, W.B.; Kavazis, A.N.; Powers, S.K. Nuclear factor-kappaB signaling contributes to mechanical ventilation-induced diaphragm weakness. Crit Care Med. 2012, 40, 927–934.
- Qiu, Y.-W.; Chen, D.; Xu, M.-Y.; Li, S.-T. Beneficial effects of dantrolene on sepsis-induced diaphragmatic dysfunction are associated with downregulation of high-mobility group box 1 and calpain-caspase-3 proteolytic pathway. J. Surg. Res. 2016, 200, 637–647.
- Files, D.C.; D’Alessio, F.R.; Johnston, L.F.; Kesari, P.; Aggarwal, N.R.; Garibaldi, B.T.; Mock, J.R.; Simmers, J.L.; DeGorordo, A.; Murdoch, J.; et al. A critical role for muscle ring finger-1 in acute lung injury-associated skeletal muscle wasting. Am. J. Respir. Crit. Care Med. 2012, 185, 825–834.
- Moarbes, V.; Mayaki, D.; Huck, L.; Leblanc, P.; Vassilakopoulos, T.; Petrof, B.J.; Hussain, S.N.A. Differential regulation of myofibrillar proteins in skeletal muscles of septic mice. Physiol. Rep. 2019, 7, e14248.
- Tang, H.; Shrager, J.B. The Signaling Network Resulting in Ventilator-induced Diaphragm Dysfunction. Am. J. Respir. Cell Mol. Biol. 2018, 59, 417–427.
- Picard, M.; Azuelos, I.; Jung, B.; Giordano, C.; Matecki, S.; Hussain, S.N.A.; White, K.; Li, T.; Liang, F.; Benedetti, A.; et al. Mechanical ventilation triggers abnormal mitochondrial dynamics and morphology in the diaphragm. J. Appl. Physiol. 2015, 118, 1161–1171.
- Dridi, H.; Yehya, M.; Barsotti, R.; Reiken, S.; Angebault, C.; Jung, B.; Jaber, S.; Marks, A.R.; Lacampagne, A.; Matecki, S. Mitochondrial oxidative stress induces leaky ryanodine receptor during mechanical ventilation. Free Radic. Biol. Med. 2020, 146, 383–391.
- Tang, H.; Lee, M.; Budak, M.T.; Pietras, N.; Hittinger, S.; Vu, M.; Khuong, A.; Hoang, C.D.; Hussain, S.N.; Levine, S.; et al. Intrinsic apoptosis in mechanically ventilated human diaphragm: Linkage to a novel Fos/FoxO1/Stat3-Bim axis. FASEB J. 2011, 25, 2921–2936.
- Supinski, G.S.; Schroder, E.A.; Callahan, L.A. Mitochondria and Critical Illness. Chest 2020, 157, 310–322.
- Azuelos, I.; Jung, B.; Picard, M.; Liang, F.; Li, T.; Lemaire, C.; Giordano, C.; Hussain, S.; Petrof, B.J. Relationship between Autophagy and Ventilator-induced Diaphragmatic Dysfunction. Anesthesiology 2015, 122, 1349–1361.
- Oliveira, T.S.; Santos, A.T.; Andrade, C.B.V.; Silva, J.D.; Blanco, N.; Rocha, N.N.; Woyames, J.; Silva, P.L.; Rocco, P.R.M.; da-Silva, W.S.; et al. Sepsis Disrupts Mitochondrial Function and Diaphragm Morphology. Front. Physiol. 2021, 12, 704044.
- Demoule, A.; Divangahi, M.; Yahiaoui, L.; Danialou, G.; Gvozdic, D.; Labbe, K.; Bao, W.; Petrof, B.J. Endotoxin triggers nuclear factor-kappaB-dependent up-regulation of multiple proinflammatory genes in the diaphragm. Am. J. Respir. Crit. Care Med. 2006, 174, 646–653.
- Huang, M.; Cai, S.; Su, J. The Pathogenesis of Sepsis and Potential Therapeutic Targets. Int. J. Mol. Sci. 2019, 20, 5376.
- Li, L.F.; Liu, Y.Y.; Chen, N.H.; Chen, Y.H.; Huang, C.C.; Kao, K.C.; Chang, C.H.; Chuang, L.P.; Chiu, L.C. Attenuation of ventilation-induced diaphragm dysfunction through toll-like receptor 4 and nuclear factor-kappaB in a murine endotoxemia model. Lab Investig. 2018, 98, 1170–1183.
- Ning, F.; Wang, X.; Shang, L.; Wang, T.; Lv, C.; Qi, Z.; Wu, D. Low molecular weight heparin may prevent acute lung injury induced by sepsis in rats. Gene 2015, 557, 88–91.
- Luan, Z.-G.; Naranpurev, M.; Ma, X.-C. Treatment of Low Molecular Weight Heparin Inhibits Systemic Inflammation and Prevents Endotoxin-Induced Acute Lung Injury in Rats. Inflammation 2014, 37, 924–932.
- Scheinhorn, D.J.; Chao, D.C.; Hassenpflug, M.S.; Gracey, D.R. Post-ICU weaning from mechanical ventilation: The role of long-term facilities. Chest 2001, 120, 482S–484S.
- Schönhofer, B.; Euteneuer, S.; Nava, S.; Suchi, S.; Kohler, D. Survival of mechanically ventilated patients admitted to a specialised weaning centre. Intensive Care Med. 2002, 28, 908–916.
- Bigatello, L.M.; Stelfox, H.T.; Berra, L.; Schmidt, U.; Gettings, E.M. Outcome of patients undergoing prolonged mechanical ventilation after critical illness. Crit. Care Med. 2007, 35, 2491–2497.
- Mauri, T.; Pivi, S.; Bigatello, L.M. Prolonged mechanical ventilation after critical illness. Minerva Anestesiol. 2008, 74, 297–301.
- Wu, Y.-K.; Kao, K.-C.; Hsu, K.-H.; Hsieh, M.-J.; Tsai, Y.-H. Predictors of successful weaning from prolonged mechanical ventilation in Taiwan. Respir. Med. 2009, 103, 1189–1195.
- Jubran, A.; Grant, B.J.B.; Duffner, L.A.; Collins, E.G.; Lanuza, D.M.; Hoffman, L.A.; Tobin, M.J. Long-Term Outcome after Prolonged Mechanical Ventilation. A Long-Term Acute-Care Hospital Study. Am. J. Respir. Crit. Care Med. 2019, 199, 1508–1516.
- Kao, K.-C.; Hu, H.-C.; Fu, J.-Y.; Hsieh, M.-J.; Wu, Y.-K.; Chen, Y.-C.; Chen, Y.-H.; Huang, C.-C.; Yang, C.-T.; Tsai, Y.-H. Renal replacement therapy in prolonged mechanical ventilation patients with renal failure in Taiwan. J. Crit. Care 2011, 26, 600–607.
- Lone, N.I.; Walsh, T.S. Prolonged mechanical ventilation in critically ill patients: Epidemiology, outcomes and modelling the potential cost consequences of establishing a regional weaning unit. Crit. Care 2011, 15, R102.
- Shih, C.-Y.; Hung, M.-C.; Lu, H.-M.; Chen, L.; Huang, S.-J.; Wang, J.-D. Incidence, life expectancy and prognostic factors in cancer patients under prolonged mechanical ventilation: A nationwide analysis of 5,138 cases during 1998–2007. Crit. Care 2013, 17, R144.
- Huang, H.-Y.; Lee, C.-S.; Chiu, T.-H.; Chen, H.H.; Chan, L.-Y.; Chang, C.-J.; Chang, S.-C.; Hu, H.-C.; Kao, K.-C.; Chen, N.-H.; et al. Clinical outcomes and prognostic factors for prolonged mechanical ventilation in patients with acute stroke and brain trauma. J. Formos. Med. Assoc. 2021, 121, 162–169.
- Angus, D.C. Caring for the critically ill patient: Challenges and opportunities. JAMA 2007, 298, 456–458.
- Brower, R.G. Consequences of bed rest. Crit. Care Med. 2009, 37, S422–S428.
- Chen, Y.-H.; Lin, H.-L.; Hsiao, H.-F.; Chou, L.-T.; Kao, K.-C.; Huang, C.-C.; Tsai, Y.-H. Effects of Exercise Training on Pulmonary Mechanics and Functional Status in Patients with Prolonged Mechanical Ventilation. Respir. Care 2012, 57, 727–734.
- Van Wetering, C.R.; Hoogendoorn, M.; Mol, S.J.; Rutten-van Molken, M.P.; Schols, A.M. Short- and long-term efficacy of a community-based COPD management programme in less advanced COPD: A randomised controlled trial. Thorax 2010, 65, 7–13.
- Ho, S.-C.; Lin, H.-C.; Kuo, H.-P.; Chen, L.-F.; Sheng, T.-F.; Jao, W.-C.; Wang, C.-H.; Lee, K.-Y. Exercise training with negative pressure ventilation improves exercise capacity in patients with severe restrictive lung disease: A prospective controlled study. Respir. Res. 2013, 14, 22.
- Huang, H.-Y.; Chou, P.-C.; Joa, W.-C.; Chen, L.-F.; Sheng, T.-F.; Lin, H.-C.; Yang, L.-Y.; Pan, Y.-B.; Chung, F.-T.; Wang, C.-H.; et al. Pulmonary rehabilitation coupled with negative pressure ventilation decreases decline in lung function, hospitalizations, and medical cost in COPD: A 5-year study. Medicine 2016, 95, e5119.
- Huang, H.-Y.; Lo, C.-Y.; Yang, L.-Y.; Chung, F.-T.; Sheng, T.-F.; Lin, H.-C.; Lin, C.-W.; Huang, Y.-C.; Chang, C.-J.; Chung, K.F.; et al. Maintenance Negative Pressure Ventilation Improves Survival in COPD Patients with Exercise Desaturation. J. Clin. Med. 2019, 8, 562.
- Chen, Y.-H.; Lin, H.-L.; Hsiao, H.-F.; Huang, C.-T.; Kao, K.-C.; Li, L.-F.; Huang, C.-C.; Tsai, Y.-H. Effects of an additional pressure support level on exercise duration in patients on prolonged mechanical ventilation. J. Formos. Med. Assoc. 2015, 114, 1204–1210.
- Dunn, H.; Quinn, L.; Corbridge, S.J.; Eldeirawi, K.; Kapella, M.; Collins, E.G. Mobilization of prolonged mechanical ventilation patients: An integrative review. Heart Lung 2017, 46, 221–233.
- Schreiber, A.F.; Ceriana, P.; Ambrosino, N.; Malovini, A.; Nava, S. Physiotherapy and Weaning From Prolonged Mechanical Ventilation. Respir. Care 2018, 64, 17–25.
- Bissett, B.; Gosselink, R.; Van Haren, F.M.P. Respiratory Muscle Rehabilitation in Patients with Prolonged Mechanical Ventilation: A Targeted Approach. Crit. Care 2020, 24, 103.
- Chen, S.; Su, C.-L.; Wu, Y.-T.; Wang, L.-Y.; Wu, C.-P.; Wu, H.-D.; Chiang, L.-L. Physical training is beneficial to functional status and survival in patients with prolonged mechanical ventilation. J. Formos. Med. Assoc. 2011, 110, 572–579.
- Clini, E.M.; Crisafulli, E.; Degli Antoni, F.; Beneventi, C.; Trianni, L.; Costi, S.; Fabbri, L.M.; Nava, S. Functional Recovery Following Physical Training in Tracheotomized and Chronically Ventilated Patients. Respir. Care 2011, 56, 306–313.
- Martin, U.J.; Hincapie, L.; Nimchuk, M.; Gaughan, J.; Criner, G.J. Impact of whole-body rehabilitation in patients receiving chronic mechanical ventilation. Crit. Care Med. 2005, 33, 2259–2265.
- Chiang, L.-L.; Wang, L.-Y.; Wu, C.-P.; Wu, H.-D.; Wu, Y.-T. Effects of Physical Training on Functional Status in Patients with Prolonged Mechanical Ventilation. Phys. Ther. 2006, 86, 1271–1281.
- Yang, P.-H.; Wang, C.-S.; Wang, Y.-C.; Yang, C.-J.; Hung, J.-Y.; Hwang, J.-J.; Wang, T.-H.; Chuang, I.-C.; Huang, M.-S. Outcome of Physical Therapy Intervention on Ventilator Weaning and Functional Status. Kaohsiung J. Med. Sci. 2010, 26, 366–372.
- Handelsman, H. Intermittent positive pressure breathing (IPPB) therapy. Health Technol. Assess. Rep. 1991, 1, 1–9.
- Chen, Y.-H.; Yeh, M.-C.; Hu, H.-C.; Lee, C.-S.; Li, L.-F.; Chen, N.-H.; Huang, C.-C.; Kao, K.-C. Effects of Lung Expansion Therapy on Lung Function in Patients with Prolonged Mechanical Ventilation. Can. Respir. J. 2016, 2016, 5624315.
- Snyder-Mackler, L.; Delitto, A.; Bailey, S.L.; Stralka, S.W. Strength of the quadriceps femoris muscle and functional recovery after reconstruction of the anterior cruciate ligament. A prospective, randomized clinical trial of electrical stimulation. J. Bone Jt. Surg. 1995, 77, 1166–1173.
- Sillen, M.J.H.; Speksnijder, C.M.; Eterman, R.A.; Janssen, P.P.; Wagers, S.S.; Wouters, E.F.M.; Uszko-Lencer, N.; Spruit, M.A. Effects of neuromuscular electrical stimulation of muscles of ambulation in patients with chronic heart failure or COPD: A systematic review of the English-language literature. Chest 2009, 136, 44–61.
- Maffiuletti, N.A. Physiological and methodological considerations for the use of neuromuscular electrical stimulation. Eur. J. Appl. Physiol. 2010, 110, 223–234.
- Rodriguez, P.O.; Setten, M.; Maskin, L.P.; Bonelli, I.; Vidomlansky, S.R.; Attie, S.; Frosiani, S.L.; Kozima, S.; Valentini, R. Muscle weakness in septic patients requiring mechanical ventilation: Protective effect of transcutaneous neuromuscular electrical stimulation. J. Crit. Care. 2012, 27, 319.e1–319.e8.
- Ferrando, A.A.; Lane, H.W.; Stuart, C.A.; Davis-Street, J.; Wolfe, R.R. Prolonged bed rest decreases skeletal muscle and whole body protein synthesis. Am. J. Physiol. Content 1996, 270, 627–633.
- Kortebein, P.; Ferrando, A.; Lombeida, J.; Wolfe, R.; Evans, W.J. Effect of 10 Days of Bed Rest on Skeletal Muscle in Healthy Older Adults. JAMA J. Am. Med. Assoc. 2007, 297, 1769–1774.
- Ambrosino, N.; Vitacca, M. The patient needing prolonged mechanical ventilation: A narrative review. Multidiscip. Respir. Med. 2018, 13, 6.


