Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Savina Apolloni | -- | 1791 | 2022-06-08 15:37:27 | | | |
| 2 | Dean Liu | Meta information modification | 1791 | 2022-06-09 04:15:57 | | | | |
| 3 | Dean Liu | Meta information modification | 1791 | 2022-06-10 05:19:17 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Apolloni, S.; Milani, M.; D'ambrosi, N. Neuroinflammation in Friedreich’s Ataxia. Encyclopedia. Available online: https://encyclopedia.pub/entry/23842 (accessed on 23 July 2026).
Apolloni S, Milani M, D'ambrosi N. Neuroinflammation in Friedreich’s Ataxia. Encyclopedia. Available at: https://encyclopedia.pub/entry/23842. Accessed July 23, 2026.
Apolloni, Savina, Martina Milani, Nadia D'ambrosi. "Neuroinflammation in Friedreich’s Ataxia" Encyclopedia, https://encyclopedia.pub/entry/23842 (accessed July 23, 2026).
Apolloni, S., Milani, M., & D'ambrosi, N. (2022, June 08). Neuroinflammation in Friedreich’s Ataxia. In Encyclopedia. https://encyclopedia.pub/entry/23842
Apolloni, Savina, et al. "Neuroinflammation in Friedreich’s Ataxia." Encyclopedia. Web. 08 June, 2022.
Copy Citation
Friedreich’s ataxia (FRDA) is a rare genetic disorder caused by mutations in the gene frataxin, encoding for a mitochondrial protein involved in iron handling and in the biogenesis of iron−sulphur clusters, and leading to progressive nervous system damage.
frataxin
microglia
astrocytes
neurons
1. Introduction
Friedreich’s ataxia (FRDA) is a recessive genetic disorder involving mainly the nervous system, caused by mutations in the gene frataxin (FXN), which is located in the centromeric region of chromosome 9q. When the most common mutation, which consists of the expansion of GAA repeats in the first FXN intron, is present in homozygosis, it leads to the decreased transcription of FXN, with pathologic loss of the protein function. The severity of the disease depends on the number of repeats, and it can be associated with progressive ataxia, weakness, and sensory deficits, with symptom onset usually occurring in childhood or adolescence. Patients often die of cardiomyopathy.
2. Neuroinflammation in FRDA
Specific pathways related to neuroinflammation are altered in the microglia, astrocytes, and myelinating glial cells in FRDA, as shown in Figure 1 and reported in detail in the following paragraphs.
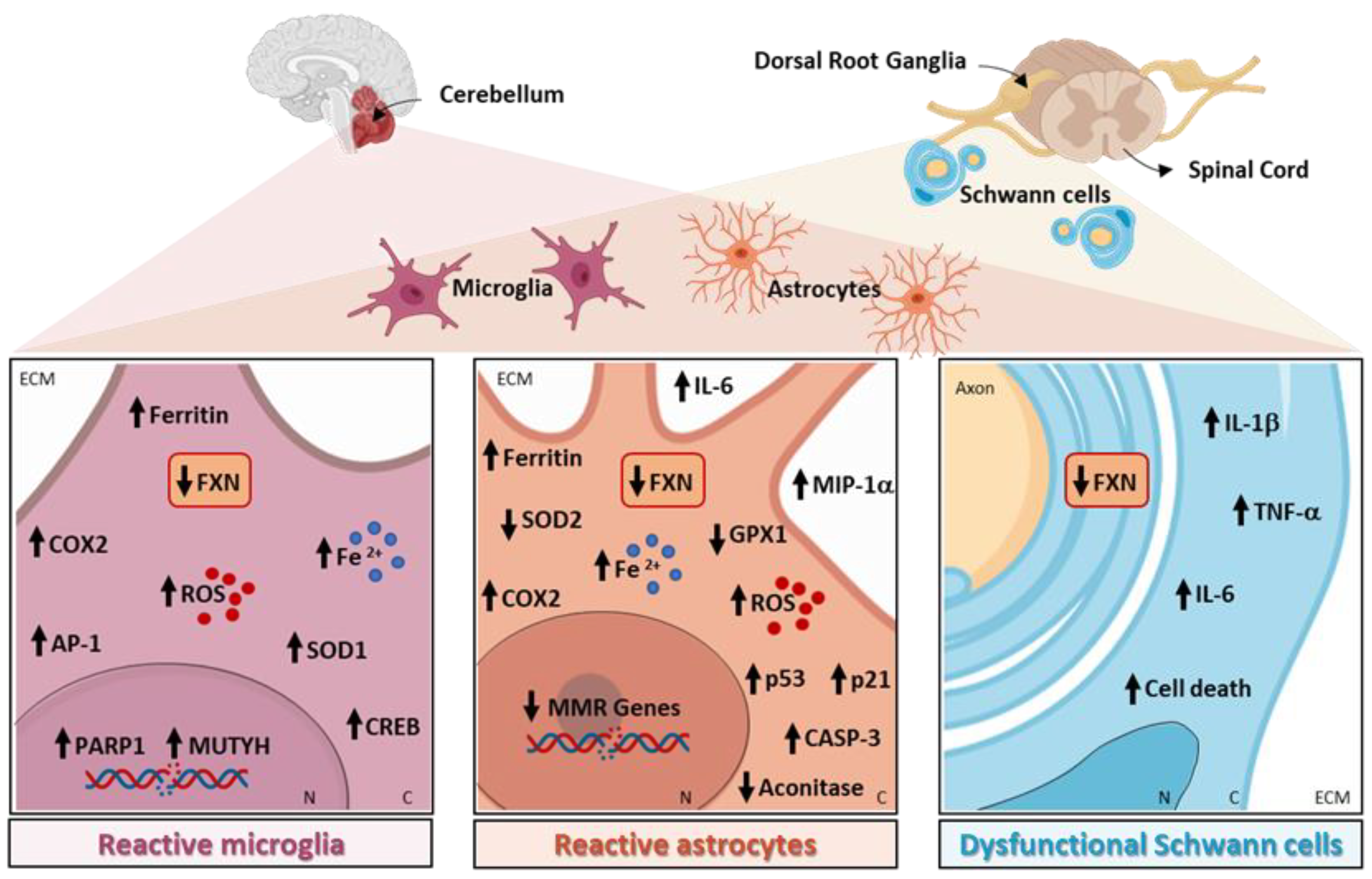
Figure 1. Neuroinflammation-related pathways altered in Friedreich’s ataxia (FRDA).
2.1. Microglia in FRDA
A confirmation of overall reactive gliosis in patients came from the brain positron emission tomography analysis, using the radioligand [18 F]-FEMPA to translocator protein (TSPO), a marker of microglia and astrocyte activation and proliferation. Using this technique, it was revealed that there was increased glial activation in the brain regions implicated in FRDA neuropathology, i.e., dentate nuclei, brainstem, superior cerebellar peduncles, and cerebellar cortex in individuals with FRDA, compared with the control subjects. The augmented binding of [18 F]-FEMPA was correlated with earlier disease onset, shorter disease duration, and an increase in plasma inflammatory cytokines, among which was interleukin-6 (IL-6) in patients with FRDA, indicating that chronic neuroinflammation could be a critical pathogenic mechanism in the disease [1].
The role of microglia activation in FRDA has been described in the KIKO mouse model of the disease, where the intracerebroventricular injection of the inflammatory stimulus lipopolysaccharide (LPS) induced a greater microglial activation compared with the healthy mice. The swollen cell bodies had shortened processes, suggested that the microglia in FRDA mice are in a more activated state than the microglia in wild-type mice. Furthermore, the authors observed an increase in oxidative damage and the DNA repair proteins MUTYH and PARP-1 in the cerebellar microglia of FRDA mice. These aberrant features were attenuated through the administration of PJ34, a PARP-1 inhibitor, suggesting that microglial PARP-1 could be an important therapeutic target in FRDA. The involvement of DNA damage in the activation of FXN-deficient microglia was further confirmed in experiments in vitro on microglial cell lines, where the knockdown of FXN increased DNA damage and the expression of the DNA repair genes MUTYH and PARP-1 [2].
In other mouse model of FRDA, YG8R, the transplantation of wild-type mouse hematopoietic stem and progenitor cells (HSPCs), resulted in the amelioration of muscle weakness and locomotor deficits [3]. In the histological analysis, sensory neurons in the dorsal root ganglia (DRGs) and mitochondria in the brain, skeletal muscle, and heart appeared intact. The authors demonstrated that, in FRDA mice, transplanted HSPCs were engrafted within the brain and spinal cord as differentiated microglia, and within the DRGs, peripheral nerves, skeletal muscle, and heart as differentiated macrophages. Hence, the robust neurological phenotypic rescue observed in HSPC-transplanted YG8R mice could be partly due to replacing the FXN-deficient microglial cells with wild-type microglia. Mechanistically, microglia transferred wild-type FXN and cyclooxygenase (COX) 8 mitochondrial proteins to neurons in vivo, suggesting the existence of a novel mechanism to be investigated in FRDA [3]. Sustaining the hypothesis that FTX deficiency leads to an increase in neuroinflammation and to the production of ROS, in the cerebella of both KIKO and YG8R mice, there was an upregulation in inducible COX2 expression and activity compared with the controls, accompanied by an increase in the transcription factors activator protein 1 (AP1) and cAMP response element-binding protein (CREB), known to drive COX2 expression. In addition, the authors showed that FXN deficiency increased the reactivity of the microglia in the cerebellum of YG8R mice after LPS treatment, further indicating an increased susceptibility to inflammation compared with the healthy mice [4].
2.2. Astrocytes in FRDA
Astrocytes play crucial roles in the pathogenesis of several forms of ataxias, where they contribute to disease progression in a phase-specific manner and represent a new target for therapeutic approaches [5]. In cerebellar tissues of FRDA patients, marked astrogliosis of the dentate nucleus is evident, as demonstrated by ferritin positive astrocytes detected near the vessel walls [6]. Moreover, the autopsy specimens of FRDA patients showed the intrusion of CNS-derived astroglia into the dorsal roots [7]. Consistently, the levels of plasma glial fibrillary acidic protein (GFAP) are significantly higher in FRDA patients, potentially reflecting glial activation [8].
Loss of FXN is detrimental not only to neurons, but also to the normal function of astroglia; cerebellar astrocytes may contribute to FRDA clinical symptoms, showing specific vulnerability to FXN deficiency [9]. In human astrocytes in vitro, the knockdown of FXN demonstrated detrimental effects to the integrity of the mitochondria, which appeared severely swollen and punctate. Accordingly, mitochondrial superoxide formation, apoptosis-related proteins p53 and p21, and activated caspase-3 were all increased. Moreover, astrocytes lacking FXN displayed abnormal secretion of several molecules, mainly associated with cell growth, immunity, and inflammation, such as IL-6 and macrophage inflammatory protein-1 alpha (MIP-1α). Remarkably, FXN-depleted astrocytes had detrimental effects on neuron development by inducing a delay in the maturation of mouse neurons and decreased neurite length and cell branching. The reduction in these features was associated with enhanced cell death, highlighting that FXN silencing in astrocytes alters their capacity to support the development of neurons. Finally, the study confirmed that the altered mitochondrial iron homeostasis in astrocytes caused by FXN deficiency leads to an increased mitochondrial iron content that favors oxidative stress and superoxide production, contributing to the non-cell-autonomous pathological process in FRDA [10].
In accordance, the astrocytes differentiated from neural stem cells obtained from the YG8R model exhibited detrimental signs, such as the reduced activity of the Fe-S containing enzyme aconitase [11]. Alteration in bioenergetic parameters is a common pathological feature of the neurodegenerative diseases leading to neuronal dysfunction, and dysfunctional aconitase, among the other bioenergetic parameters, is a crucial factor that could promote neurodegeneration [12]. In addition, FXN-deficient astrocytes showed a reduced expression of the antioxidant enzymes SOD2 and Gpx1, resulting in increased sensitivity to oxidative stress, together with a significant reduction in the expression of several DNA mismatch repair enzymes compared with the control cells [11]. Finally, FXN knockdown increased the production of ROS in the primary mouse astrocytes [13].
Sustaining a non-cell-autonomous toxic effect of FXN in vivo, the ablation of FXN in astrocytes during development in FGKO mice (where FXN is ablated in a time-dependent manner) caused severe ataxia and early death, inducing growth and survival impairments. In contrast, the mice in which FXN was knocked out in astrocytes later in life did not give rise to apparent neurological phenotypes, indicating that developing cerebellar astrocytes are more vulnerable to the lack of FXN, and suggesting a role of astrocytes in the progression of the disease [14]. Extensive neuroinflammation has been observed in YG8R mice, where FXN loss induced increased satellite cell proliferation, extensive astrocytosis, and an influx of inflammatory OX42 (CD11b/c)-positive cells in both the spinal cord and cerebellar dentate nucleus [15]. Consistently, in the KIKO mouse model, a substantial increase in astrocytosis was detected following LPS injection in the cerebellum of FRDA compared with non-transgenic mice, suggesting an increased vulnerability to inflammation, as observed for the microglia [2].
Targeting astrocytes in models of the disease represents a promising strategy. Indeed, in FGKO and YG8R mice, treatment with insulin-like growth factor I (IGF-I), previously shown to normalize FXN levels in FXN-deficient neurons and astrocyte cultures through the Akt/mTOR signaling pathway [13], proved beneficial effects for rescuing astrocyte-associated cerebellar defects and atrophy, together with the improvement of motor performances and increment in survival [14].
In addition, in YG8R mice, treatment with granulocyte-colony stimulating factor (G-CSF) and stem cell factor (SCF) markedly reduced the extent of astrocytosis and inflammatory cell infiltration within the dorsal columns, spinocerebellar, and corticospinal tracts. The results indicate that attenuating neuroinflammation slows down the progression of the disease. The neuroprotective action of this combined treatment was indeed exerted at a clinical level, resulting in a significant improvement in motor coordination and locomotor activity, even after the onset of neurological symptoms [15].
It has been demonstrated that targeting Sonic Hedgehog (SHH) with the Smoothened antagonist (SAG) rescued mitochondrial dysfunction and reverted the neurotoxicity induced by the lack of FXN in human astrocytes in vitro, showing the potential of pharmacologically targeting astrocytes cells to attenuate neurodegeneration in FRDA [16].
In Drosophila melanogaster, FXN knockdown in the glia affects fly locomotion, increases brain vacuolization due to cellular degeneration, and induces defects in lipid metabolism and oxidative stress [17], suggesting a role for these cells in the pathology. The expression of Glaz, one of the Drosophila homologs of apolipoprotein D (ApoD), in the glia of FXN-deficient flies, was sufficient to increase the lifespan and improve locomotor activity, likely because of its modulation of lipid composition and oxidation [18]. Interestingly, a genetic screen in the same model identified Drosophila mitofusin (Marf), a gene involved in mitochondrial fusion and degradation, as lying at the interface between the mitochondria and endoplasmic reticulum, a critical mediator of the pathology in the glia. Marf downregulation fully rescued some of the essential phenotypes induced by FXN silencing in the glia, such as locomotor dysfunction, brain degeneration, and lipid dyshomeostasis in the brain [19].
Overall, these results demonstrate that astrocyte activation could exacerbate or even cause neuronal dysfunctions, triggering a further amplification of astrogliosis in a detrimental vicious circle.
2.3. Myelinating Glial Cells in FRDA
Finally, myelinating glial cells are also implicated in FRDA, with oligodendroglia and Schwann cells being highly susceptible to FXN deficiency. In the dentate nucleus of FRDA patients, ferritin is expressed mainly in the oligodendrocytes, while, as the disease progresses and neurons begin to undergo atrophy, these cells disappear, being replaced by positive ferritin microglia [6]. As demonstrated in vitro, a significant decrease in the proliferation of both oligodendrocytes and Schwann cells occurred after FXN knockdown through the activation of the inflammatory pathways. Indeed, in FXN-deficient Schwann cells, the microarrays analysis showed a decrease in antioxidant genes and a substantial increase in inflammatory genes, such as IL-1β, IL-1α, IL-6, NFκB1, and Tumor Necrosis factor, confirmed at both the mRNA and protein levels, suggesting that the inflammatory cytokines produced by these cells may contribute to DRG neuron loss [20].
Altogether, this evidence suggests that the expression of mutant FXN in glial cells may act as a trigger, responsible for their reprogramming and functional impairment, contributing to the degeneration of nearby neurons.
References
- Khan, W.; Corben, L.A.; Bilal, H.; Vivash, L.; Delatycki, M.B.; Egan, G.F.; Harding, I.H. Neuroinflammation in the Cerebellum and Brainstem in Friedreich Ataxia: An -FEMPA PET Study. Mov. Disord. 2022, 37, 218–224.
- Kemp, K.C.; Cerminara, N.; Hares, K.; Redondo, J.; Cook, A.J.; Haynes, H.R.; Burton, B.R.; Pook, M.; Apps, R.; Scolding, N.J.; et al. Cytokine Therapy-Mediated Neuroprotection in a Friedreich’s Ataxia Mouse Model. Ann. Neurol. 2017, 81, 212–226.
- Cerrato, V. Cerebellar Astrocytes: Much More Than Passive Bystanders In Ataxia Pathophysiology. J. Clin. Med. 2020, 9, 757.
- Hayashi, G.; Shen, Y.; Pedersen, T.L.; Newman, J.W.; Pook, M.; Cortopassi, G. Frataxin Deficiency Increases Cyclooxygenase 2 and Prostaglandins in Cell and Animal Models of Friedreich’s Ataxia. Hum. Mol. Genet. 2014, 23, 6838–6847.
- Sandi, C.; Sandi, M.; Jassal, H.; Ezzatizadeh, V.; Anjomani-Virmouni, S.; Al-Mahdawi, S.; Pook, M.A. Generation and Characterisation of Friedreich Ataxia YG8R Mouse Fibroblast and Neural Stem Cell Models. PLoS ONE 2014, 9, e89488.
- Koeppen, A.H.; Michael, S.C.; Knutson, M.D.; Haile, D.J.; Qian, J.; Levi, S.; Santambrogio, P.; Garrick, M.D.; Lamarche, J.B. The Dentate Nucleus in Friedreich’s Ataxia: The Role of Iron-Responsive Proteins. Acta Neuropathol. 2007, 114, 163–173.
- Franco, C.; Genis, L.; Navarro, J.A.; Perez-Domper, P.; Fernandez, A.M.; Schneuwly, S.; Torres Alemán, I. A Role for Astrocytes in Cerebellar Deficits in Frataxin Deficiency: Protection by Insulin-like Growth Factor I. Mol. Cell Neurosci. 2017, 80, 100–110.
- Rocca, C.J.; Goodman, S.M.; Dulin, J.N.; Haquang, J.H.; Gertsman, I.; Blondelle, J.; Smith, J.L.M.; Heyser, C.J.; Cherqui, S. Transplantation of Wild-Type Mouse Hematopoietic Stem and Progenitor Cells Ameliorates Deficits in a Mouse Model of Friedreich’s Ataxia. Sci. Transl. Med. 2017, 9, eaaj2347.
- Harding, I.H.; Lynch, D.R.; Koeppen, A.H.; Pandolfo, M. Central Nervous System Therapeutic Targets in Friedreich Ataxia. Hum. Gene Ther. 2020, 31, 1226–1236.
- Khodagholi, F.; Shaerzadeh, F.; Montazeri, F. Mitochondrial Aconitase in Neurodegenerative Disorders: Role of a Metabolism- Related Molecule in Neurodegeneration. Curr. Drug Targets 2018, 19, 973–985.
- Navarro, J.A.; Ohmann, E.; Sanchez, D.; Botella, J.A.; Liebisch, G.; Molto, M.D.; Ganfornina, M.D.; Schmitz, G.; Schneuwly, S. Altered Lipid Metabolism in a Drosophila Model of Friedreich’s Ataxia. Hum. Mol. Genet. 2010, 19, 2828–2840.
- Franco, C.; Fernández, S.; Torres-Alemán, I. Frataxin Deficiency Unveils Cell-Context Dependent Actions of Insulin-like Growth Factor I on Neurons. Mol. Neurodegener. 2012, 7, 51.
- Shen, Y.; McMackin, M.Z.; Shan, Y.; Raetz, A.; David, S.; Cortopassi, G. Frataxin Deficiency Promotes Excess Microglial DNA Damage and Inflammation That Is Rescued by PJ34. PLoS ONE 2016, 11, e0151026.
- Smith, F.M.; Kosman, D.J. Molecular Defects in Friedreich’s Ataxia: Convergence of Oxidative Stress and Cytoskeletal Abnormalities. Front. Mol. Biosci. 2020, 7, 569293.
- Vicente-Acosta, A.; Giménez-Cassina, A.; Díaz-Nido, J.; Loria, F. The Smoothened Agonist SAG Reduces Mitochondrial Dysfunction and Neurotoxicity of Frataxin-Deficient Astrocytes. J. Neuroinflamm. 2022, 19, 93.
- Loría, F.; Díaz-Nido, J. Frataxin Knockdown in Human Astrocytes Triggers Cell Death and the Release of Factors That Cause Neuronal Toxicity. Neurobiol. Dis. 2015, 76, 1–12.
- Navarro, J.A.; Botella, J.A.; Metzendorf, C.; Lind, M.I.; Schneuwly, S. Mitoferrin Modulates Iron Toxicity in a Drosophila Model of Friedreich’s Ataxia. Free Radic. Biol. Med. 2015, 85, 71–82.
- Zeitlberger, A.M.; Thomas-Black, G.; Garcia-Moreno, H.; Foiani, M.; Heslegrave, A.J.; Zetterberg, H.; Giunti, P. Plasma Markers of Neurodegeneration Are Raised in Friedreich’s Ataxia. Front. Cell Neurosci. 2018, 12, 366.
- Edenharter, O.; Schneuwly, S.; Navarro, J.A. Mitofusin-Dependent ER Stress Triggers Glial Dysfunction and Nervous System Degeneration in a Drosophila Model of Friedreich’s Ataxia. Front. Mol. Neurosci. 2018, 11, 38.
- Lu, C.; Schoenfeld, R.; Shan, Y.; Tsai, H.-J.; Hammock, B.; Cortopassi, G. Frataxin Deficiency Induces Schwann Cell Inflammation and Death. Biochim. Biophys. Acta 2009, 1792, 1052–1061.
More
Information
Subjects:
Pathology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.6K
Entry Collection:
Neurodegeneration
Revisions:
3 times
(View History)
Update Date:
10 Jun 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No