Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ho Lee | -- | 3372 | 2022-05-18 08:28:06 | | | |
| 2 | Conner Chen | Meta information modification | 3372 | 2022-05-18 09:30:03 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Lee, H. Obesity-Associated Cancers. Encyclopedia. Available online: https://encyclopedia.pub/entry/23049 (accessed on 27 July 2026).
Lee H. Obesity-Associated Cancers. Encyclopedia. Available at: https://encyclopedia.pub/entry/23049. Accessed July 27, 2026.
Lee, Ho. "Obesity-Associated Cancers" Encyclopedia, https://encyclopedia.pub/entry/23049 (accessed July 27, 2026).
Lee, H. (2022, May 18). Obesity-Associated Cancers. In Encyclopedia. https://encyclopedia.pub/entry/23049
Lee, Ho. "Obesity-Associated Cancers." Encyclopedia. Web. 18 May, 2022.
Copy Citation
Obesity, one of the major problems in modern human society, is correlated with various diseases, including type 2 diabetes mellitus (T2DM). In particular, epidemiological and experimental evidence indicates that obesity is closely linked to at least 13 different types of cancer. The mechanisms that potentially explain the link between obesity and cancer include hyperactivation of the IGF pathway, metabolic dysregulation, dysfunctional angiogenesis, chronic inflammation, and interaction between pro-inflammatory cytokines, endocrine hormones, and adipokines.
obesity
obesity-associated cancer
mouse cancer model
1. Investigation of the Link between Obesity and Pancreatic Cancer
Epidemiological studies have revealed that pancreatic cancer is caused by obesity and that pancreatic cancer is a high-risk factor for developing diabetes [1]. The most common malignancy of the exocrine pancreas is pancreatic ductal adenocarcinoma (PDAC), which comprises over 90% of cases. Oncogenic KRAS mutations are the most common mutation in PDAC patients, with a frequency exceeding 90% [2]. In addition, tumor suppressors such as CDKN2A, TP53, and SMAD4 are mutated in greater than 95%, 50–70%, and 55% of PDAC patients, respectively. Even though revolutionary cancer therapeutics have been developed, including combination chemotherapy, small molecule-mediated targeted therapy, and immunotherapies, the survival rate of PDAC patients is still low, at about 10%.
1.1. Kras-Driven PDAC in a Mouse Model
The KPC mouse is one of the most-studied GEMMs of PDAC and considered the most appropriate surrogate model of human PDAC in terms of clinical and histological characteristics [3] (Table 1). Hingorani and colleagues have generated the KPC model with endogenous control and concomitant expression of KrasG12D and Trp53R172H in the pancreas [4]. This concomitant expression of oncogenic Kras and Trp53 shortens the time of tumor progression from pancreatic intraepithelial neoplasia (PanIN) lesions to metastatic adenocarcinoma. In the model, spontaneous cancer can cause metastasis in the lung and liver about 2.5 months after birth. Although the KPC mouse is a successful model for the study of PDAC, it still has some limitations, such as a limited impact on the progression of other diseases, such as diabetes and infrequent signs of para-neoplastic diabetes [5]. From this point of view, the KC mouse may be a more useful model in obesity studies. These transgenic animals express the oncogenic KrasG12D alleles in acinar cells of the pancreas and show preneoplastic PanIN lesions that ultimately progress to invasive and metastatic PDAC [4]. However, invasive and metastatic cancer development in the mice is relatively slow, with PanIN-3 lesions detected at 4–6 months and full-blown metastatic cancer after 12 months of age. Another GEMM of PDAC, the KPP mouse, expresses the KrasG12D oncogenes and involves the loss of a functional Pten gene in the pancreas [6]. KPP mice show a progressive loss of skeletal and adipose mass due to pancreatic tumors and also display a similar gene ontology in the muscle to cachectic patients. Therefore, KPPC mice are regarded as a useful model in the preclinical study of cachexia. Recently, Collins and colleagues developed a new mouse model of PDAC, the iKras model (Ptf1a-Cre;LSL-rtTA;LSL-Trp53R172H;TetO-KrasG12D) [7]. In this mouse, KrasG12D is inducible, which leads to reversible and pancreas-specific expression of the oncogenic Kras gene. Compared with the KPC model, iKras model mice have a mean survival of about seven months, more rapid formation of PDAC, and metastasis with a larger tumor mass.
Table 1. Mouse models commonly used in studies on obesity-cancer.
| Name | Mutations or Transgenes | Obesity Phenotypes | Cancer or Disease Phenotypes |
|---|---|---|---|
| KPC | KrasG12D;Trp53R172H; Pdx1-Cre [4] |
HDFs promote desmoplasia [8] | Limited impact on the progression of other diseases, such as diabetes, due to rapid tumor progression |
| KC | KrasG12D; Pdx1-Cre [4] |
More useful model in obesity studies than KPC mouse | Slow progression in invasive and metastatic cancer |
| KPP | KrasG12D; Pten(f/f);Ptf1a-CreERT [6] |
Preclinical model of cachexia | Displaying a similar gene ontology in muscle to cachectic patients |
| APCMin | Stop codon at codon 850 [9][10] | HFDs increase systemic and local inflammation before the onset of obesity [11][12] | Developing more than 100 intestinal polyps, but rare invasive adenocarcinoma |
| Apc1638N | Insertion of the neomycin cassette in codon 1638 [9][10] | HFDs accelerate tumor development and increased multiplicity [13] | Close resemblance to human CRC |
| MMTV-PyMT | HDFs enhance primary tumorigenesis and metastasis [14][15] | Similar features with the luminal subtype of human breast cancer | |
| MMTV-neu | HFDs effect, controversial [16][17] | Similar features with the luminal subtype of human breast cancer | |
| MMTV-TGFα | HFDs increase adiposity and shorten mammary tumor latency | Useful model of postmenopausal human breast cancer. | |
| MMTV-Wnt1 | CR * inhibit transplanted tumors in postmenopausal model [18] | Features of the luminal and basal types. | |
| C3(1)-Tag | HFDs increase tumor initiation [19][20] | Similar features with the basal subtype of human breast cancer |
* CR, calorie restriction.
1.2. Study of Obesity Using Kras-Driven PDAC Models
Several studies using these Kras-driven PDAC models reported that a HFD promotes the development of pancreatic cancer. Accordingly, subsequent studies aimed at identifying the relationship between obesity and pancreatic cancer and the specific mechanisms involved are ongoing. Analysis of KC mice subjected to a HFD revealed that chronic inflammation and autophagy dysregulation play a role in diet-induced pancreatic cancer progression and genetic alteration, contributing to mortality [21]. Ob/ob mice have been crossed with KC mice to model obesity-associated pancreatic cancer, generating KCO mice. KCO mice are obese and show an increased primary ductal tumor burden, enhanced PDAC progression, and a dramatically shortened lifespan compared with non-obese KC mice [22]. These phenotypes were more severe than those in HFD models. This experiment also revealed that obesity promotes tumorigenesis independent of new driver mutations, such as Trp53, Cdkn2a/p16, and Smad4 mutations, frequent in PDAC. To verify whether the inflammation induced by a HFD and the accompanying upregulation of cyclooxygenase-2 (Cox2) increase PDAC progression, KC mice were crossed with Cox2 conditional knockout mice, generating Cox2flox/flox;LSL-KrasG12D;Ela-CreERT [23]. A HFD increases Kras activity, fibrotic stroma, the numbers of PanIN lesions, and PDAC incidence (Figure 1). Despite the dramatic increase in the number of PanIN lesions, inflammation, and fibrosis in LSL-KrasG12D;Ela-CreERT mice fed a HFD, there was no evidence for an increase in these phenotypes in Cox2flox/flox;LSL-KrasG12D;Ela-CreERT mice. In addition, the administration of Cox2 inhibitor to KC mice prevented the tumorigenic effects of the HFD. These results indicate that HFDs can activate oncogenic Kras via Cox2, causing inflammation and fibrosis in the pancreas and tumor development.
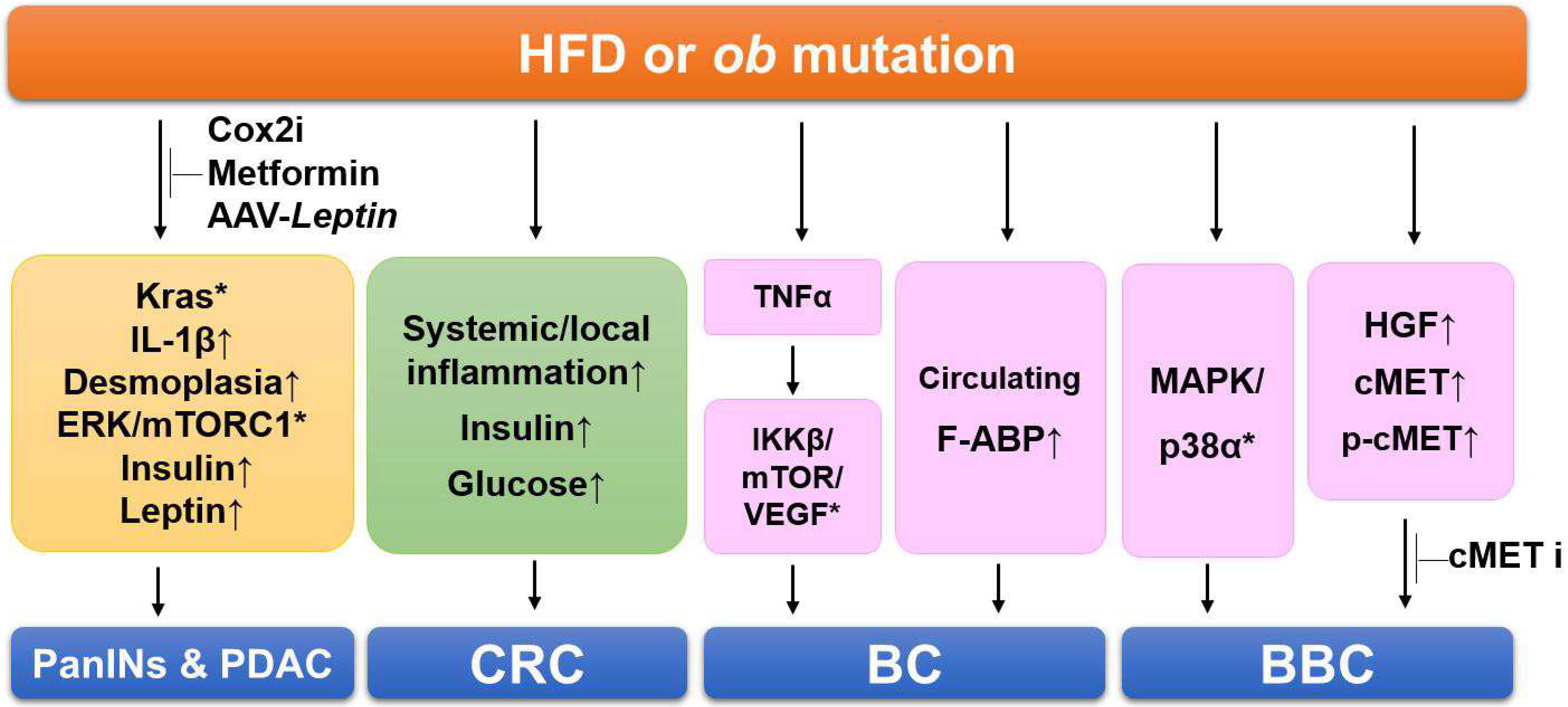
Figure 1. Summary of the signaling pathways involving obesity-linked cancers in mouse models. HFDs cause Kras activation, inflammation, fibrosis, and increased insulin and leptin levels, resulting in PDAC progression. Administration of Cox2 inhibitor (Cox2i), metformin, and AAV-Leptin prevents HDF-induced and manifestations in PDAC models. HDFs increase systemic and local inflammation, elevate insulin and glucose levels in the colorectal cancer (CRC) models. In the breast cancer (BC) models, obesity increases several genes related to proliferation and the serum levels of A-FABP. HFDs activate multiples kinases of MAPK/p38 and increase hepatocyte growth factor (HGF) secretion in basal-like breast cancer (BBC) models. Administration of cMET inhibitor (cMETi) prevents tumor development in BBC-bearing mice. Upward-pointing arrows, increased levels of indicated proteins/adipokines; *, activation of indicated signaling pathways.
Desmoplasia is a key feature in pancreatic cancer associated with accelerated tumor growth and the impaired delivery of anti-cancer drugs due to reduced perfusion [2]. Obesity is known to promote inflammation and fibrosis in the normal pancreas. An experiment with KPC and iKras mice revealed that desmoplasia is promoted by a HFD in the models [8]. Adipocyte-secreted IL-1β recruits tumor-associated neutrophils, which activate pancreatic stellate cells. Consistent with these observations, depletion of tumor-associated neutrophils, inhibition of IL-1β, or inactivation of pancreatic stellate cells prevents the rapid obesity-mediated progression of PDAC. These results suggest that aggravation of desmoplasia is a critical mechanism of obesity-promoted PDAC progression and that clinically available anti-fibrotic or inflammatory agents could be used to treat PDAC in obese patients.
To investigate whether anti-diabetic drugs affect PDAC development, Kras-driven cancer models have been subjected to a HFD and/or metformin, a widely used anti-diabetic drug [24]. Metformin administration prevented HDF-induced manifestations, including increased body weight, hepatic steatosis, depleted intact acini, PanIN lesion formation, and activation of ERK and mTORC1 in the pancreas (Figure 1). Metformin also normalized the HFD-induced hyperinsulinemia and hyperleptinemia and completely abrogated the HFD-induced further increase in PDAC incidence. Based on these results, an anti-diabetic drug could be a novel chemopreventive drug for PDAC.
To verify the effect of weight loss on tumorigenesis, KCO mice, including an ob mutation, have been chosen because the obesity phenotype can be rapidly reversed through leptin restoration [22]. An adeno-associated virus-Leptin (AAV-Leptin) leads to sustained leptin secretion in mice through a single intramuscular injection. Multiple phenotypes of leptin deficiency, including obesity, hyperglycemia, and subfertility, can be reversed by AAV-Leptin administration (Figure 1). Interestingly, AAV-Leptin administration before advanced tumor development delays tumor progression proportional to the degree of body weight loss. However, AAV-Leptin administration after advanced tumor development causes loss of body weight without affecting tumorigenesis and survival. The study showed that islet cholecystokinin is aberrantly expressed in response to obesity, promoting oncogenic Kras-driven pancreatic ductal tumorigenesis. These results suggest that an obesity-associated microenvironment change drives PDAC progression and implicates endocrine–exocrine signaling beyond insulin in PDAC development.
2. Investigation of the Link between Obesity and Colorectal Cancer (CRC)
Experimental results indicate that obesity is a critical risk factor for colon cancer [25] and that colon cancer risk can be reduced by preventing or reversing obesity through a calorie-restricted diet. Investigation and identification of these associations’ molecular mechanisms will help to reveal novel therapeutic targets and develop approaches for treating obesity-related CRC. For this purpose, several mouse models of colon cancer have been used in the context of obesity, including Apc mutant mice and the azoxymethane (AOM)-treated models [11][12][26][27][28].
2.1. Mouse Models of CRC
Carcinogen-induced mouse models of CRC were first developed more than 80 years ago. Since then, considerable numbers of animal models have been generated to investigate tumorigenesis in the colon [9][10][29]. GEMMs of intestinal/colon cancer have the same mutation found in human patients and show similar pathogeneses to sporadic and inherited human colon cancers, dependent on the activation or inactivation of specific molecular pathways (Table 1). One of the earliest GEMMs of colon cancer is the ApcMin mouse (Min = multiple intestinal neoplasias), which was developed in 1990 in the laboratory of William Dove [30] and has been widely used for over 30 years in various fields. The ApcMin mouse includes a germline mutation in the Apc gene, an autosomal dominant loss-of-function Apc gene caused by a stop codon at codon 850 generated by exposure to N-ethyl-N-nitrosourea (ENU), a highly potent carcinogen. In each animal, heterozygous ApcMin mice develop more than 100 intestinal polyps, mainly located in the small intestine. The mutation most frequently found in human colon cancer patients is that in adenomatous polyposis coli (APC); this mutation is observed in over 80% of cases. Therefore, ApcMin mice have commonly been used as the model of the human familial adenomatous polyposis (FAP) syndrome. Several additional models with Apc mutations have been generated in subsequent work, most of which have truncating mutations in the gene [9][10]. The Apc∆716 mouse was generated by inserting a neomycin cassette into Apc codon 716, generating a truncated peptide (~80 kDa). This mutation causes many adenomas, mainly in the small intestine, and a reduced number in the large intestine and fewer extra-intestinal lesions, similar to ApcMin mouse. The Apc1638N mouse was generated by inserting a neomycin cassette into Apc codon 1638. These animals develop 5–6 intestinal adenomas per animal and exhibit a broad spectrum of extra-intestinal manifestations, including cutaneous cysts and multifocal desmoids. Although Apc1638N mice develop intestinal tumors, the mice are less tumorigenic, have a longer latency than ApcMin mice, and develop invasive adenocarcinoma, which is rarely found in ApcMin, as well as splenomegaly and desmoid formation. Given these characteristics, the mice are considered to resemble human CRC more closely. AOM is an alkylating agent that generates free radicals. Administration of AOM to C57BL/6 mice causes mutations in β-catenin and tumor formation with low incidence [31]. AOM treatment of ApcMin mice increases the incidence of colon cancer and the number of polyps, but tumor formation predominantly occurs in the small intestine
2.2. Investigation of the Link between Obesity and Colon Cancer
The number of polyps increases and the spontaneous development of multiple intestinal neoplasias in ApcMin mice is accelerated by a HFD [11][26]. In addition to an increase in intestinal polyp formation in the ApcMin mice, exposure to a HFD increases systemic and local inflammation before the onset of obesity, as well as metabolic syndrome-associated characteristics, including an elevated level of insulin or glucose [11][12] (Figure 1). AOM-induced CRC models have also examined if diet-induced obesity affects colon cancer progression [27][28]. In the experiment, male C57BL/6J mice fed regular chow or a HFD for eight weeks were administered AOM to cause colon cancers. The mice were classified into four groups: regular chow, HFD, regular chow switched to a HFD, and a HFD switched to regular chow. The results indicated that colon cancer development is promoted by prior HFD-induced obesity, even without weight gain and HFD maintenance, meaning that colon cancer can be promoted by obesity itself, without the additional effects of a HFD or genetic alteration.
Mouse cancer models have also tested the effects of dietary components on obesity-related cancer. For example, walnuts mitigate cancer risk and include various bioactive components with anti-inflammatory and antioxidant effects. Therefore, walnuts have the potential to protect or counteract pathways that initiate or drive the development of obesity-related tumors. Using C57BL/6J mice, Apc1638N, ApcΔ14, and MC38 colon cancer cells, researchers have investigated the effects of walnuts on intestinal homeostasis and tumor growth or attenuation in the context of HFDs and obesity [13]. The intake of walnuts dramatically protects against intestinal tumor growth and progression and preserves the function of intestinal stem cells in the context of HFDs and obesity. These results are additional evidence that some diets (e.g., walnuts) have the potential to break the obesity–colon cancer link.
Obesogenic conditions during the intrauterine and nursing periods could affect obesity and CRC. It has been examined whether obesogenic conditions in various life periods influence obesity and intestinal tumorigenesis in adult ApcMin mice [32][33]. The results found that exposure to an obesogenic condition in utero and during the nursing period (45% fat) caused an increase in body weight in both control and ApcMin mice and increased the number of intestinal polyps in ApcMin offspring as adults compared with a control diet (10% fat). These results indicate that obesogenic conditions in the intrauterine and nursing periods could be critical for susceptibility to dietary fat-induced obesity and intestinal tumor development.
3. Investigation of the Link between Obesity and Breast Cancer
Obesity is associated with an approximately 40% increased risk of breast cancer recurrence and death, resulting in poor survival outcomes [34]. In particular, obesity strongly correlates with breast cancer risk in postmenopausal women [35][36] and is found in about 50% of all breast cancer cases in older women [37]. This correlation is established in estrogen receptor-positive breast cancer but is less well established in human epidermal growth factor receptor 2-positive and triple-negative subtypes. Although several mechanisms have been proposed for the link between obesity and cancer, the detailed and established molecular mechanisms of obesity-linked breast tumorigenesis remain elusive.
3.1. Mouse Models of Breast Cancer
To generate GEMMs of breast cancer, several oncogenes have been targeted in the mammary gland, such as Myc, PyMT, Her2/neu, and Hras, using mouse mammary tumor virus long terminal repeat (MMTV-LTR) or whey acidic protein (Wap) promoters [38][39][40][41][42]. Based on molecular profiling of mammary tumors from mouse models of breast cancer, individual tumors were clustered together on a dendrogram, suggesting that some models (MMTV-neu, MMTV-PyMT, Wap-Myc, and Wap-Int3) showed similar features to the luminal subtype of human breast cancer and that other models (C3(1)-Tag, Wap-Tag, and Brca1-deficient models) were similar to the basal subtype (Table 1).
MMTV-PyMT mice express polyomavirus middle T antigen driven by MMTV-LTR, restricting its expression to the mammary epithelium. This mouse model shows a high penetrance of early-onset mammary cancer than other mammary tumor models. In particular, palpable mammary tumors developed in MMTV-PyMT females could metastasize to the lung. MMTV-neu mice express the activated rat neu gene via the MMTV-LRT promoter. In the activated rat neu oncogene, a valine is replaced by glutamic acid at amino acid 664. Tumors formed in the mice are multifocal and stochastic, and adenocarcinoma is usually detected in older mice (about 23 weeks after birth). Based on the gene expression profile and the high expression of XBP1, a human luminal subtype-defining gene, the model is considered to represent the human luminal subtype [38][43]. MMTV-TGFα transgenic mice express human TGFα cDNA under the control of the MMTV-LTR [44][45]. The transgenic mice show abnormal phenotypes, including lobular hyperplasia, cystic hyperplasia, adenoma, and adenocarcinoma in the mammary epithelium. In addition, the mice show slow development of breast cancer, with onset at 12 months after birth, representing a useful model of postmenopausal human breast cancer. MMTV-Wnt1 transgenic mice express Wnt1 driven by MMTV-LTR, developing breast cancer that shows luminal and basal subtype features [46]. The transgenic construct of C3(1)-Tag mice includes the simian virus 40 early-region transforming sequences driven by the regulatory element of the rat prostate steroid-binding protein C3(1) gene. Most transgenic mice (~95%) show features of the human basal-like subtype, such as high proliferation and high expression of keratins 5 and 17 and P-cadherin. The female mice usually develop mammary intraepithelial neoplasia similar to ductal carcinoma in situ (DCIS) by three months after birth and subsequently develop mammary adenocarcinoma by six months of age with 100% penetrance. In addition, about 10–15% of female mice develop lung metastasis.
3.2. Investigation of the Link between Obesity and Breast Cancer
For determining the effects of obesity on the initiation and progression of breast cancer, two mouse models of obesity have been used: one is an orthotopic mammary tumor-bearing genetic model (ob/ob mice), while the other is an orthotopic mammary tumor-bearing diet-induced obesity model [47]. The results showed that the initiation and development of breast cancer were promoted by obesity in the models and that the expression was increased of several genes related to proliferation, including TNFa, VEGF, IKKβ, and mTOR, indicating that TNFα may activate the IKKβ/mTOR/VEGF signaling pathway in the tumors of obese mice (Figure 1). Other groups have also reported that the initiation of breast cancer and lung metastasis was increased by a HFD containing soybean oil in MMTV-PyMT mice [14][15]. Furthermore, they identified the upregulation of pro-inflammatory cytokines, adipokines, and angiogenic factors, which were closely associated with the increased and aggressive features of mammary tumors induced by the HFD. From these studies, a molecular interpretation may explain why the incidence of breast cancer is higher and the prognosis is worse in obese women.
To investigate whether a dietary intervention that reduced adiposity before tumor onset would reverse HFD-induced breast cancer, breast cancer models (C3(1)-Tag mice) were fed a low-fat diet (LFD) or HFD and then an obese group exposed to a HFD was switched to a LFD to cause weight loss [19][20]. Weight loss before tumor initiation inhibited the increase in HFD-induced cancer, which reduced tumor latency and preneoplastic lesions, including atypical ductal hyperplasia and DCIS. Kinome analysis explained that multiple kinases upstream of MAPK/p38α were activated by the HFD-induced weight gain and reversed by the weight loss (Figure 1). These may be novel targets in obesity-associated breast cancer, particularly basal-like breast cancer (BBC). Finally, these results suggested that tumor initiation can be promoted by the HFD-exposed microenvironment, which was reprogrammed by weight loss and the restoration of a lean phenotype. Another study of C3(1)-Tag mice determined that obesity significantly increases the secretion of hepatocyte growth factor (HGF) and decreases the latency of breast cancer [48] (Figure 1). The expression of HGF, cMET, and phospho-cMET in the normal mammary glands was elevated, accelerating BBC tumor progression compared with lean controls. In addition, weight loss significantly reversed the HFD-induced effects on latency and the activation of HGF/cMET signaling in normal mammary tissue and cMET in normal mammary and breast cancer tissue. Because obesity leads to a critical elevation in HGF/cMET secretion in breast cancer, researchers have investigated whether a small-molecule cMET inhibitor (e.g., crizotinib) has effects on BBC tumor progression in LFD- and HFD-fed C3(1)-Tag BBC mice [49]. cMET inhibitor was administered before tumor development or at an early stage. It significantly decreased tumor volume by 27.96% and 37.29% and average cancer vascularity by 35.04% and 33.52% in the LFD- and HFD-fed C3(1)-Tag mice, respectively. These results suggest that tumor development and microvascular density in basal-like tumor-bearing mice could be inhibited by cMET inhibition with crizotinib.
Recently, more and more studies have reported that metabolic changes are strongly associated with the development of breast cancer. For example, the serum levels of A-FABP are dramatically increased in obesity [45][50], which is associated with the stemness and aggressive development of breast and ovarian cancer (Figure 1). From these results, circulating A-FABP could be a new link between obesity and breast cancer and a new potential therapeutic target to treat obesity-associated tumors.
References
- Andersen, D.K.; Korc, M.; Petersen, G.M.; Eibl, G.; Li, D.; Rickels, M.R.; Chari, S.T.; Abbruzzese, J.L. Diabetes, Pancreatogenic Diabetes, and Pancreatic Cancer. Diabetes 2017, 66, 1103–1110.
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220.
- Lee, J.W.; Komar, C.A.; Bengsch, F.; Graham, K.; Beatty, G.L. Genetically Engineered Mouse Models of Pancreatic Cancer: The KPC Model (LSL-Kras (G12D/+); LSL-Trp53 (R172H/+); Pdx-1-Cre), Its Variants, and Their Application in Immuno-oncology Drug Discovery. Curr. Protoc. Pharmacol. 2016, 73, 14.39.1–14.39.20.
- Siveke, J.T.; Schmid, R.M. Chromosomal instability in mouse metastatic pancreatic cancer—It’s Kras and Tp53 after all. Cancer Cell 2005, 7, 405–407.
- Pasquale, V.; Dugnani, E.; Liberati, D.; Marra, P.; Citro, A.; Canu, T.; Policardi, M.; Valla, L.; Esposito, A.; Piemonti, L. Glucose metabolism during tumorigenesis in the genetic mouse model of pancreatic cancer. Acta Diabetol. 2019, 56, 1013–1022.
- Talbert, E.E.; Cuitino, M.C.; Ladner, K.J.; Rajasekerea, P.V.; Siebert, M.; Shakya, R.; Leone, G.W.; Ostrowski, M.C.; Paleo, B.; Weisleder, N.; et al. Modeling Human Cancer-induced Cachexia. Cell Rep. 2019, 28, 1612–1622.e4.
- Collins, M.A.; Bednar, F.; Zhang, Y.; Brisset, J.C.; Galban, S.; Galban, C.J.; Rakshit, S.; Flannagan, K.S.; Adsay, N.V.; Pasca di Magliano, M. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J. Clin. Investig. 2012, 122, 639–653.
- Incio, J.; Liu, H.; Suboj, P.; Chin, S.M.; Chen, I.X.; Pinter, M.; Ng, M.R.; Nia, H.T.; Grahovac, J.; Kao, S.; et al. Obesity-Induced Inflammation and Desmoplasia Promote Pancreatic Cancer Progression and Resistance to Chemotherapy. Cancer Discov. 2016, 6, 852–869.
- Fodde, R.; Smits, R.; Clevers, H. APC, signal transduction and genetic instability in colorectal cancer. Nat. Rev. Cancer 2001, 1, 55–67.
- Jackstadt, R.; Sansom, O.J. Mouse models of intestinal cancer. J. Pathol. 2016, 238, 141–151.
- Day, S.D.; Enos, R.T.; McClellan, J.L.; Steiner, J.L.; Velazquez, K.T.; Murphy, E.A. Linking inflammation to tumorigenesis in a mouse model of high-fat-diet-enhanced colon cancer. Cytokine 2013, 64, 454–462.
- Dazard, J.E.; Sandlers, Y.; Doerner, S.K.; Berger, N.A.; Brunengraber, H. Metabolomics of ApcMin/+ mice genetically susceptible to intestinal cancer. BMC Syst. Biol. 2014, 8, 72.
- Guan, F.; Tabrizian, T.; Novaj, A.; Nakanishi, M.; Rosenberg, D.W.; Huffman, D.M. Dietary Walnuts Protect Against Obesity-Driven Intestinal Stem Cell Decline and Tumorigenesis. Front. Nutr. 2018, 5, 37.
- Cowen, S.; McLaughlin, S.L.; Hobbs, G.; Coad, J.; Martin, K.H.; Olfert, I.M.; Vona-Davis, L. High-Fat, High-Calorie Diet Enhances Mammary Carcinogenesis and Local Inflammation in MMTV-PyMT Mouse Model of Breast Cancer. Cancers 2015, 7, 1125–1142.
- Sundaram, S.; Yan, L. High-fat Diet Enhances Mammary Tumorigenesis and Pulmonary Metastasis and Alters Inflammatory and Angiogenic Profiles in MMTV-PyMT Mice. Anticancer Res. 2016, 36, 6279–6287.
- Cleary, M.P.; Grande, J.P.; Juneja, S.C.; Maihle, N.J. Diet-induced obesity and mammary tumor development in MMTV-neu female mice. Nutr. Cancer 2004, 50, 174–180.
- Ecker, B.L.; Lee, J.Y.; Sterner, C.J.; Solomon, A.C.; Pant, D.K.; Shen, F.; Peraza, J.; Vaught, L.; Mahendra, S.; Belka, G.K.; et al. Impact of obesity on breast cancer recurrence and minimal residual disease. Breast Cancer Res. 2019, 21, 41.
- Nogueira, L.M.; Dunlap, S.M.; Ford, N.A.; Hursting, S.D. Calorie restriction and rapamycin inhibit MMTV-Wnt-1 mammary tumor growth in a mouse model of postmenopausal obesity. Endocr. Relat. Cancer 2012, 19, 57–68.
- Qin, Y.; Sundaram, S.; Essaid, L.; Chen, X.; Miller, S.M.; Yan, F.; Darr, D.B.; Galanko, J.A.; Montgomery, S.A.; Major, M.B.; et al. Weight loss reduces basal-like breast cancer through kinome reprogramming. Cancer Cell Int. 2016, 16, 26.
- Tiwari, P.; Blank, A.; Cui, C.; Schoenfelt, K.Q.; Zhou, G.; Xu, Y.; Khramtsova, G.; Olopade, F.; Shah, A.M.; Khan, S.A.; et al. Metabolically activated adipose tissue macrophages link obesity to triple-negative breast cancer. J. Exp. Med. 2019, 216, 1345–1358.
- Chang, H.H.; Moro, A.; Takakura, K.; Su, H.Y.; Mo, A.; Nakanishi, M.; Waldron, R.T.; French, S.W.; Dawson, D.W.; Hines, O.J.; et al. Incidence of pancreatic cancer is dramatically increased by a high fat, high calorie diet in KrasG12D mice. PLoS ONE 2017, 12, e0184455.
- Chung, K.M.; Singh, J.; Lawres, L.; Dorans, K.J.; Garcia, C.; Burkhardt, D.B.; Robbins, R.; Bhutkar, A.; Cardone, R.; Zhao, X.; et al. Endocrine-Exocrine Signaling Drives Obesity-Associated Pancreatic Ductal Adenocarcinoma. Cell 2020, 181, 832–847.e18.
- Philip, B.; Roland, C.L.; Daniluk, J.; Liu, Y.; Chatterjee, D.; Gomez, S.B.; Ji, B.; Huang, H.; Wang, H.; Fleming, J.B.; et al. A high-fat diet activates oncogenic Kras and COX2 to induce development of pancreatic ductal adenocarcinoma in mice. Gastroenterology 2013, 145, 1449–1458.
- Chang, H.H.; Moro, A.; Chou, C.E.N.; Dawson, D.W.; French, S.; Schmidt, A.I.; Sinnett-Smith, J.; Hao, F.; Hines, O.J.; Eibl, G.; et al. Metformin Decreases the Incidence of Pancreatic Ductal Adenocarcinoma Promoted by Diet-induced Obesity in the Conditional KrasG12D Mouse Model. Sci. Rep. 2018, 8, 5899.
- Ye, P.; Xi, Y.; Huang, Z.; Xu, P. Linking Obesity with Colorectal Cancer: Epidemiology and Mechanistic Insights. Cancers 2020, 12, 1408.
- van Kranen, H.J.; van Iersel, P.W.; Rijnkels, J.M.; Beems, D.B.; Alink, G.M.; van Kreijl, C.F. Effects of dietary fat and a vegetable-fruit mixture on the development of intestinal neoplasia in the ApcMin mouse. Carcinogenesis 1998, 19, 1597–1601.
- Tuominen, I.; Al-Rabadi, L.; Stavrakis, D.; Karagiannides, I.; Pothoulakis, C.; Bugni, J.M. Diet-induced obesity promotes colon tumor development in azoxymethane-treated mice. PLoS ONE 2013, 8, e60939.
- Park, S.Y.; Kim, J.S.; Seo, Y.R.; Sung, M.K. Effects of diet-induced obesity on colitis-associated colon tumor formation in A/J mice. Int. J. Obes. 2012, 36, 273–280.
- Burtin, F.; Mullins, C.S.; Linnebacher, M. Mouse models of colorectal cancer: Past, present and future perspectives. World J. Gastroenterol. 2020, 26, 1394–1426.
- Moser, A.R.; Pitot, H.C.; Dove, W.F. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 1990, 247, 322–324.
- De Robertis, M.; Massi, E.; Poeta, M.L.; Carotti, S.; Morini, S.; Cecchetelli, L.; Signori, E.; Fazio, V.M. The AOM/DSS murine model for the study of colon carcinogenesis: From pathways to diagnosis and therapy studies. J. Carcinog. 2011, 10, 9.
- Ngo, H.T.; Hetland, R.B.; Steffensen, I.L. The intrauterine and nursing period is a window of susceptibility for development of obesity and intestinal tumorigenesis by a high fat diet in Min/+ mice as adults. J. Obes. 2015, 2015, 624023.
- Steffensen, I.L. Obesity and Intestinal Tumorigenesis in Adult Min/+ Mice from Early-life High-fat Diet Exposure Were Not Inherited Transgenerationally. Anticancer Res. 2016, 36, 3871–3882.
- Jiralerspong, S.; Goodwin, P.J. Obesity and Breast Cancer Prognosis: Evidence, Challenges, and Opportunities. J. Clin. Oncol. 2016, 34, 4203–4216.
- Ogden, C.L.; Carroll, M.D.; Kit, B.K.; Flegal, K.M. Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA 2014, 311, 806–814.
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014, 384, 766–781.
- Goodwin, P.J.; Stambolic, V. Impact of the obesity epidemic on cancer. Annu. Rev. Med. 2015, 66, 281–296.
- Vargo-Gogola, T.; Rosen, J.M. Modelling breast cancer: One size does not fit all. Nat. Rev. Cancer 2007, 7, 659–672.
- Guy, C.T.; Cardiff, R.D.; Muller, W.J. Induction of mammary tumors by expression of polyomavirus middle T oncogene: A transgenic mouse model for metastatic disease. Mol. Cell Biol. 1992, 12, 954–961.
- Guy, C.T.; Webster, M.A.; Schaller, M.; Parsons, T.J.; Cardiff, R.D.; Muller, W.J. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc. Natl. Acad. Sci. USA 1992, 89, 10578–10582.
- Sinn, E.; Muller, W.; Pattengale, P.; Tepler, I.; Wallace, R.; Leder, P. Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: Synergistic action of oncogenes in vivo. Cell 1987, 49, 465–475.
- Park, M.K.; Lee, C.H.; Lee, H. Mouse models of breast cancer in preclinical research. Lab. Anim. Res. 2018, 34, 160–165.
- Gruvberger, S.; Ringner, M.; Chen, Y.; Panavally, S.; Saal, L.H.; Borg, A.; Ferno, M.; Peterson, C.; Meltzer, P.S. Estrogen receptor status in breast cancer is associated with remarkably distinct gene expression patterns. Cancer Res. 2001, 61, 5979–5984.
- Cleary, M.P.; Grossmann, M.E.; Ray, A. Effect of obesity on breast cancer development. Vet. Pathol. 2010, 47, 202–213.
- Hao, J.; Zhang, Y.; Yan, X.; Yan, F.; Sun, Y.; Zeng, J.; Waigel, S.; Yin, Y.; Fraig, M.M.; Egilmez, N.K.; et al. Circulating Adipose Fatty Acid Binding Protein Is a New Link Underlying Obesity-Associated Breast/Mammary Tumor Development. Cell Metab. 2018, 28, 689–705.e5.
- Tsukamoto, A.S.; Grosschedl, R.; Guzman, R.C.; Parslow, T.; Varmus, H.E. Expression of the int-1 gene in transgenic mice is associated with mammary gland hyperplasia and adenocarcinomas in male and female mice. Cell 1988, 55, 619–625.
- Chen, C.T.; Du, Y.; Yamaguchi, H.; Hsu, J.M.; Kuo, H.P.; Hortobagyi, G.N.; Hung, M.C. Targeting the IKKbeta/mTOR/VEGF signaling pathway as a potential therapeutic strategy for obesity-related breast cancer. Mol. Cancer Ther. 2012, 11, 2212–2221.
- Sundaram, S.; Freemerman, A.J.; Johnson, A.R.; Milner, J.J.; McNaughton, K.K.; Galanko, J.A.; Bendt, K.M.; Darr, D.B.; Perou, C.M.; Troester, M.A.; et al. Role of HGF in obesity-associated tumorigenesis: C3(1)-TAg mice as a model for human basal-like breast cancer. Breast Cancer Res. Treat. 2013, 142, 489–503.
- Cozzo, A.J.; Sundaram, S.; Zattra, O.; Qin, Y.; Freemerman, A.J.; Essaid, L.; Darr, D.B.; Montgomery, S.A.; McNaughton, K.K.; Ezzell, J.A.; et al. cMET inhibitor crizotinib impairs angiogenesis and reduces tumor burden in the C3(1)-Tag model of basal-like breast cancer. Springerplus 2016, 5, 348.
- Hotamisligil, G.S.; Bernlohr, D.A. Metabolic functions of FABPs--mechanisms and therapeutic implications. Nat. Rev. Endocrinol. 2015, 11, 592–605.
More
Information
Subjects:
Cell Biology; Endocrinology & Metabolism
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
919
Revisions:
2 times
(View History)
Update Date:
18 May 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No