Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Andrea Bianconi | -- | 3318 | 2022-04-20 11:27:16 | | | |
| 2 | Rita Xu | Meta information modification | 3318 | 2022-04-20 11:53:04 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Bianconi, A.; , .; Rizzo, F.; Salvati, L.; Zeppa, P.; Cofano, F. Tumor Microenvironment in Glial Neoplasm. Encyclopedia. Available online: https://encyclopedia.pub/entry/21995 (accessed on 25 June 2026).
Bianconi A, , Rizzo F, Salvati L, Zeppa P, Cofano F. Tumor Microenvironment in Glial Neoplasm. Encyclopedia. Available at: https://encyclopedia.pub/entry/21995. Accessed June 25, 2026.
Bianconi, Andrea, , Francesca Rizzo, Luca Salvati, Pietro Zeppa, Fabio Cofano. "Tumor Microenvironment in Glial Neoplasm" Encyclopedia, https://encyclopedia.pub/entry/21995 (accessed June 25, 2026).
Bianconi, A., , ., Rizzo, F., Salvati, L., Zeppa, P., & Cofano, F. (2022, April 20). Tumor Microenvironment in Glial Neoplasm. In Encyclopedia. https://encyclopedia.pub/entry/21995
Bianconi, Andrea, et al. "Tumor Microenvironment in Glial Neoplasm." Encyclopedia. Web. 20 April, 2022.
Copy Citation
Despite the multidisciplinary management in the treatment of glioblastomas, the average survival of GBM patients is still 15 months. In recent years, molecular biomarkers have gained more and more importance both in the diagnosis and therapy of glial tumors. At the same time, it has become clear that non neoplastic cells, which constitute about 30% of glioma mass, dramatically influence tumor growth, spread, and recurrence. This is the main reason why, in recent years, scientific research has been focused on understanding the function and the composition of tumor microenvironment and its role in gliomagenesis and recurrence.
glioma
microenvironment
GAMs
microglia
1. Introduction
Despite the multidisciplinary management in the treatment of glioblastoma, the average survival is 15 months [1]. In recent years, molecular biomarkers have gained more and more importance both in the diagnosis and therapy of glial tumors [2]. Non neoplastic cells are believed to constitute up to 30% of glioma mass and are responsible for the creation of the so-called tumor microenvironment (TME) [3][4], which includes specialized glioma stem cells (GSCs), glioma cells, stromal cells including resident glial cells and immune cells such as monocytes, tumor-associated macrophages/microglia (TAMs), and lymphocytes [5]. Intra- and inter-tumor heterogeneity is therefore mainly related to the peritumor microenvironment. Understanding which type of cell constitutes the majority of TME, what their role is, and to what extent they can influence the development and spread of gliomas assumes great importance, especially for therapeutic implications, in order to focus pharmacological research on a specific cellular target. TME dramatically influences tumor growth, spread, and recurrence; for example, TAMS acts directly to promote glioblastoma growth and to create an immunosuppressive TME. Recent evidence indicates how cancer cells can subvert the peritumor microenvironment, promoting angiogenesis, invasion, and metastasis [6]. Understanding how glioma cells interact with cells in the microenvironment is an essential step in understanding disease progression.
The activation of microglial cells and the recruitment of circulating monocytes in the brain is mainly induced by chemokines, neurotransmitters, ligands of complement receptors, and a new mode of communication by the release of extracellular vesicles (EVs). Considering the large number of studies present in the literature regarding the role of the tumor microenvironment in gliomagenesis, the goal of the entry is to summarize the current evidence concerning the TME, better understanding TAM composition, and highlighting the mechanisms of interaction between microglia and tumor cells, as this could have implications both for therapeutic and prognostic aims.
2. Differentiation and Role of Resident Microglia and Circulating Monocytes
In human gliomas, TME is constituted by a mix of two different cell types: microglia, the resident macrophages of CNS, and monocytes, which originate from hematopoietic stem cells (HSCs) and travel through the blood to reach brain when blood–brain barrier (BBB) is damaged. These cells not only have two ontogenetically different origins, but also express some different surface receptors and have, perhaps, different mechanisms of action in supporting gliomas growth and spread. (Figure 1).
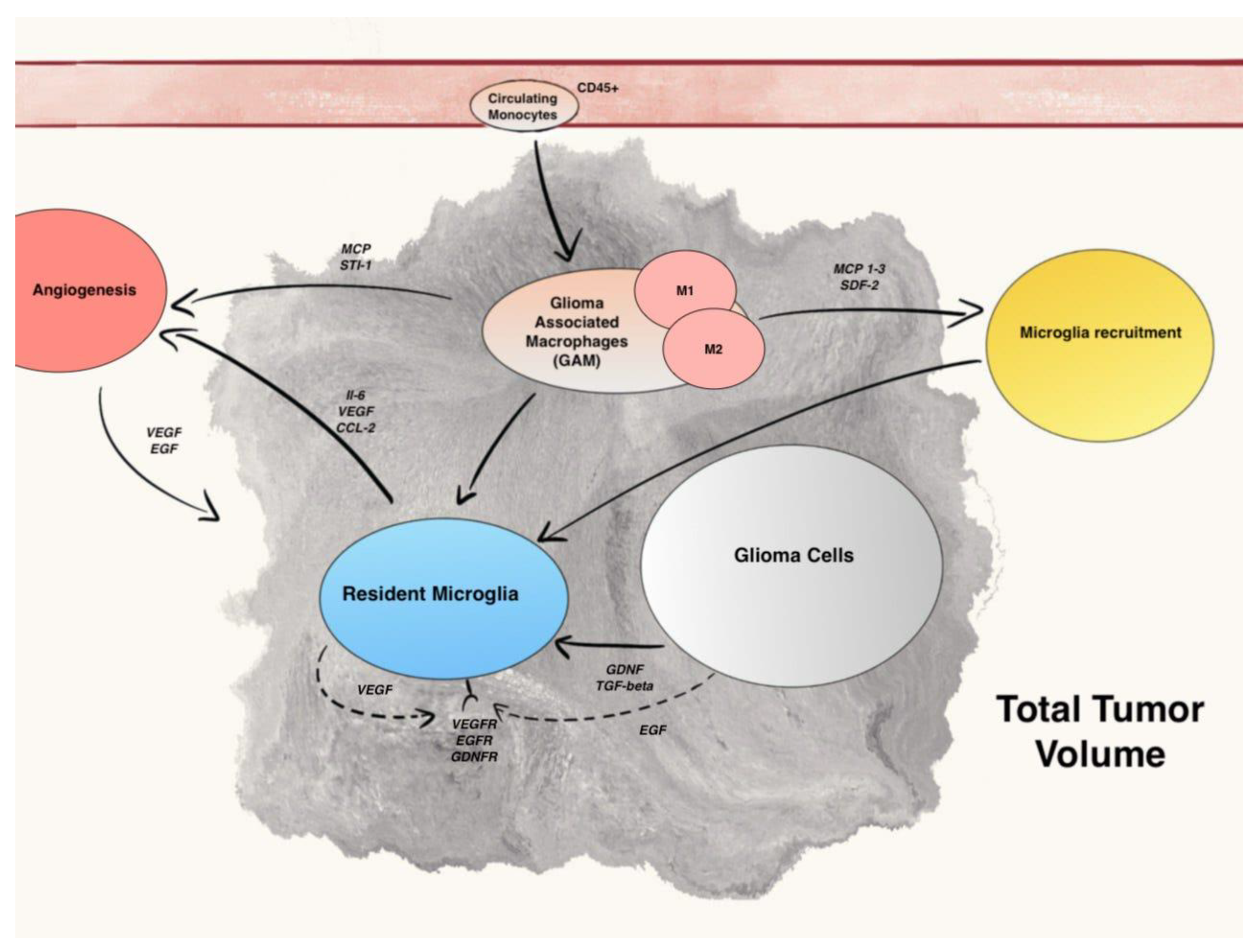
Figure 1. Summary of main communication pathways between GAM, microglia, and Glioma cells.
Normal brain tissue is colonized exclusively by quiescent microglia. In the context of microglia cells differentiation in normal brain tissue, a recent study demonstrated the existence of different microglia subsets by using a single-cell-based immune phenotyping by time-of-flight mass cytometry (CyTOF) and single-cell RNA sequencing (scRNA-seq) [7]. Conducted on 4396 microglial cells from control brain regions of 15 adults, in this study, the authors identified eight major clusters (C1–C9, with the exclusion of C4) within the microglia cloud. C2 subset was characterized by the strong expression of major histocompatibility complex II (MHC-II) and antiviral immunity genes, such as HLA-DRA, CD74, and IFI44L; C3 subset was associated with the high expression of CX3CR1 and TMEM119; C6 and C7 subsets were characterized by low expression of CX3CR1 and high expression of integrin-receptor-binding protein and metabolism genes, including SPP1, APOE, and LPL. These clusters are therefore involved in antigen processing and presentation of peptide antigen. The remaining smaller clusters C1, C5, C8, and C9, associated with the expression of chemokine and cytokine genes, including CCL2 and IL1B, are instead involved in positive regulation of chemotaxis and MAPK cascade. In the same study, an association between brain regions and specific microglia clusters was demonstrated. In fact, C2 and C5–C7 were overexpressed in white matter microglia, while C3 and C8 were overexpressed in gray matter microglia [7]. Under pathological conditions, microglia density increased through active recruitment of bone marrow-derived microglial progenitors from the bloodstream and, at the same time, HSCs infiltrate brain through chemoattraction [8][9][10].
The distinction between microglia and invading monocytes has always been made using anti-CD45 antibody, with microglial cells expressing low CD45 (CD45low) and HSCs with a high expression of CD45 (CD45high). Different studies, based on sampling human glioma by fluorescent-activated cell sorting, demonstrated that the majority of glioma associated macrophages and monocytes (GAM) are actually HSCs. A more recent study, instead based on irradiation chimeras, showed that resident microglia is the most represented member of GAM [11]. Another study, based exclusively on CX3CR1 or CCR2 antibodies, demonstrated again the prevalence of HSCs in TAM (CCR2+ CX3C1−) and the relatively low presence of resident macrophages (CCR2− CX3C1+) [12]. In 2020, Friebel et al., by using a combination of experimental approach, successfully identified the origin of each TAM population stemming either from CNS-resident microglia or HSCs. They confirmed that glioma TME is mostly characterized by TAM of microglial origin, but, interestingly enough, they also found a major invasion of HSCs in patient with IDH1 wild type gliomas in comparison to IDH mutated gliomas [13].
The lack of consensus in the literature, at least partly determined by the different methods of cell sorting (surface antibody or chimeras), underlines the necessity of new organized studies, in order to better understand TAM composition and to assess if targeting these cells can have a prognostic significance in gliomas treatment.
3. Microglia Recruitment Factors
Tumorigenesis is not only driven by genetic alterations and function of tumoral cells, but is also highly influenced by the tumor microenvironment, composed by cellular and non-cellular elements. In brain tumors, especially gliomas, tumor stroma is mainly composed of immune cells such as resident microglia and circulating macrophages and monocytes.
The activation of microglial cells and the recruitment of circulating monocytes (HSCs) in the brain is mainly induced by chemokines, neurotransmitters, and ligands of complement receptors.
One of the first chemoattractant identified was monocyte chemoattractant protein-1 (MCP-1), also known as CCL2. MCP-1 role in tumor growth has been studied in rats, where tumors generated with CCL2-expressing cells were more than three-fold larger in size than control-transfected tumors [14]. Additionally, it has been observed that in vitro chemoattraction could successfully be inhibited by an MCP-1-neutralizing antibody, while in an experiment conducted in vivo in mice, glioma cell-derived MCP-1 increased GAM infiltration. MCP-1 expression has also been associated with glioma grade [14], neoangiogenesis, tumor cells proliferation, and invasiveness [15].
However, a more recent study enhanced the importance of monocyte chemotactic protein 3 (MCP-3) role in microglia and HSCs recruitment over MCP-1 in human gliomas [16].
In a murine astrocytoma model, tumor secreted stroma-derived factor-1 (SDF-1) has been found responsible for microglia and macrophage recruitment, especially in hypoxic areas of brain [17].
Chemokine CX3CL1 (also known as fractalkine or neurotactin) exists as a membrane-anchored protein and as a potentially soluble form. The highly specific receptor for CX3CL1, called CX3CR1, is expressed by microglial cells in normal brain tissue by endothelial cells and platelets [18]. In a study conducted on GAM, CX3CR1 was overexpressed at both mRNA and protein level, and it was found that CX3CL1 promoted their adhesion and migration in vitro, through the induction of matrix metalloproteases (MMPs) 2, 9, and 14 [19]. Conversely, a more recent study demonstrated that loss of the fractalkine receptor CX3CR1 in microglia and monocytes in glioma-bearing mice resulted in increased tumor incidence and shorter survival, but did not affect the accumulation of microglia/macrophages in peri-tumoral areas. According to the authors, the first effect is related to the upregulation of interleukin 1β (IL-1β) expression in CX3CR1-deficient microglia macrophages and monocytes. IL-1β, indeed, has been found responsible for tumor growth and infiltration of cancer stem cells phenotype, both in mice and human Proneural glioma stem cells [20].
These discordant data suggest that the role of fractalkine in gliomagenesis and its therapeutic implications has yet to be defined.
Growth factor glial cell-derived neurotrophic factor (GDNF), secreted by murin and human glioma cells, is a strong microglia attractant in vitro, while having a small role in glioma-induced astrogliosis [21].
CSF-1, another soluble factor released by glioma cells, can serve as chemoattractant in vivo. In mice treated with CSF-1R antagonist, in fact, a reduction in GAM density and of glioblastoma invasiveness was observed [22]. CSF-1 is also responsible for microglia polarization into the pro-tumorigenic M2-like phenotype [23].
The knockdown of granulocyte -macrophage colony-stimulating factor (GM-CSF) in brain slices reduced microglia-macrophages invasion and the growth of intracranial gliomas in vivo [24].
Epidermal growth factor (EGF), whose receptor (EGFR) is expressed on microglial cells, is responsible for GAM migration and motility in in vitro experiments in a dose-dependent way. Moreover, as EGF is secreted by activated microglia itself in vivo, it may act as an autocrine modulator and amplifier of microglial cells functions.
In conclusion, several soluble factors, secreted by both glioma and microglial cells, can act as GAM chemoattractant in glioma bearing brain. Therefore, the inhibition of these ligands and/or their receptors could represent a new frontier in the research for therapeutic strategies in glioma patients.
4. The M1–M2 Polarization of GAM
Different pathological stimuli can activate quiescent resident microglia, such as injuries in brain trauma, tumors, and viral and bacterial infections [25]. Immunogenic antigens such as lipopolysaccharide (LPS) use several receptors, including Toll-like receptors (TLRs), nucleotide-binding oligomerization domain-(NOD)-like receptors, and scavenger receptors (SRs), to induce pathogens clearance through phagocytosis and, in the end, to trigger an immune response inside the central nervous system (CNS) [26]. Via signal transducer and activator of transcription (STAT-1), LPS can also activate the expression of pro-inflammatory cytokines, such as IL-1α, IL-6, IL-12, and IL-23, and chemokines, such as CC-chemokine ligand (CCL)2–5 and CCL8–11. Redox molecules, as NADPH-ossidase and inducible nitric oxide synthase (iNOS) for nitric oxide production, are also involved in the creation of a metabolic state called pro-inflammatory M1-phenotype [27][28].
Microglial cells expand and amplificate this activation by expressing co-stimulatory molecules (CD 40, CD80, and CD86) and exposing high levels of major histocompatibility complex (MHC) II molecules on their surface [27][29][30], serving as antigen presenting cells (APC). Periphery monocytes can be activated by resident microglia through tumor necrosis factor (TNF)-α.
On the other side, an anti-inflammatory M2-like phenotype is simultaneously activated in order to prevent tissue damages, to downregulate immune response, and to lead to CNS recovery.
Different molecules have been identified as M2 polarized microglia/macrophages markers, such as CD163 (scavenger receptor), CD204, and CD206 (macrophage mannose receptor) [31][32].
A further distinction inside M2 polarized cells has been proposed, with three categories, M2a, M2b, and M2c, that are believed to develop in different conditions and to be triggered by various environmental stimuli [33]. These categories exert distinct functions in the contest of anti-inflammatory response.
T-helper cell secretion of IL-4 is responsible for triggering a M2a polarization. The activation of IL4-Receptor (IL-4R) on microglia-macrophages surface, indeed, induces the production, via STAT 6 pathway, of anti-inflammatory cytokines and chemokines such as transforming growth factor (TGF)-β and CCL15, -17, -22, and -24 [34][35]. Simultaneously, IL-4R signaling leads to a silencing of the M1-characteristic NF-κB signaling [27].
M2b subtype is responsible for secretion of IL-1, IL-6, IL-10, CCL1, and TNF-α, via TLRs or IL-1R antagonists [36]. M2b activation leads, eventually, to immune regulation through TH2 and regulatory T cell (Treg) activation.
M2a and M2b subtypes play similar roles in immune regulation: the first can stimulate Th2 response and induce pathogens killing and allergies development, while the second is involved in Th2 response and immune regulation.
M2c subtype, instead, is a major promoter of tumor growth as, in neoplastic conditions, it is responsible for the attenuation of inflammatory responses, tissue remodeling, and matrix deposition [36].
Tumor cells are responsible for production and secretion of IL-10, TGF-β, and glucocorticoids; in particular, IL-10, through STAT3 signal, leads to a rise in TGF-β and peroxisome proliferator-activated receptor (PPAR)-γ expression and, in the end, to the activation of anti-inflammatory response [27].
M2c phenotype facilitates extracellular matrix (ECM) deposition and tissue remodeling by the expression of versican, antitrypsin, and pentraxin 3 [37][38].
For more than twenty years, M2-polarization of TAM has been a cornerstone of the solid tumors knowledge [39][40]. The immunosuppressive environment, also responsible for tissue remodeling and angiogenesis, has been linked to tumor progression [36] and poor prognosis [41]. This paradigm, however, has been challenged in the last few years, especially in brain tumors, where the role of GAM polarization and the immune system in tumor development, spread, and recurrence seems to be more complicated than previously thought [20][42][43].
The first major difficulty is linked to the scarceness of distinct markers for M1 and M2 polarized cells. Surface receptors such as CD68 and cytoplasmic protein IBA1 are considered general microglia/macrophages markers (pan-M/M markers) [32].
Markers such as CD163, CD204, CD206, arginase 1 (ARG1), FIZZ1, and phosphorylated STAT3 (pSTAT3) have been attributed to M2-polarized GAMs, while CD40, CD74, and MHC II have been linked to M1-polarized GAMs [27].
Komohara et al. demonstrated that glioma cell-derived factors, such as TGF-β and macrophage colony-stimulating factor (M-CSF), promote the upregulation of several M2 markers, such as CD163 and CD204, and therefore M2-polarization of GAM [40].
Different studies have been focused on the correlation between survival in patients with high grade glioma and the concomitant expression of either M1 or M2 polarization markers in GAM.
In one study, the analysis of GAM composition showed the predominance of M2 polarization, with a major expression of CD163+ and CD204+ cells, and its association with a worse prognosis [40].
In another study, CD74, a M1-polarization marker, was found to be expressed in GAM and was positively related to longer patient survival [44].
However, this mutually exclusive state of polarization of GAM is true in vitro, but it is now clear that this model cannot represent the complexity of in vivo setting, as different studies demonstrated that GAM can express both M1 and M2 markers at the same time [45][46][47]. Moreover, with the secretion of a variety of soluble factors, glioma cells can induce both M1 and M2 polarization in GAM. Glioma-derived M-CSF and TGF-beta shift microglia and macrophages phenotype towards a M2-type and are responsible for tumor growth [23][40]. In the same way, glioma cells can inhibit M1 phenotype with the activation of mTor and CSF-1 [48]. Other studies have suggested that M1 specific markers or associated pathways positively correlate with glioma growth, such as glioma-derived IL1-β [20].
A more recent study, focused on the prognostic significance of M1/M2 polarization GAM, demonstrated a mixed polarization phenotype, with parallel expression of presumptive M1 and M2 markers, without an univocal state of polarization.
As already mentioned before, Sankowski et al. demonstrated that SPP1 gene, encoding for a proinflammatory cytokine called osteopontine, is more expressed in C6 and C7 clusters of normal brain microglia, especially in people aged over 50 years. Afterwards, they compared osteopontine expression in GBM samples to control the white matter of four subjects >50 years of age, and found that the percentage of IBA1+SPP1+ cells was more than double in GBM samples compared with controls, demonstrating an over-expression of SPP1 gene in the GAM clusters and the similarity of GAM phenotype with the one of aging-associated microglia [7].
In the same study, with the application of the StemID2 algorithm, a gradual downregulation of microglial homeostatic genes (including CX3CR1 and CSF1R) and a concomitant upregulation of inflammatory and metabolism genes (including TREM2 and APOE) was observed in glioma associated microglia.
In conclusion, Sankowski et al. demonstrated the heterogeneity of GAM and its time-related progression from an homeostatic phenotype to a proinflammatory and metabolic phenotype, with lower expression of the microglial core signature and higher expression of proinflammatory genes such as SPP1 and several type I interferon genes, including APOE and CD163, complicating the picture of GAM expression in human glioma even further [7].
Even though the dual M1/M2 model is an oversimplification of in vivo GAM activation, it is still a useful model to find therapeutic target and to direct further tumor environment characterization.
5. Microglia Regulation of Glioma Growth, Progression, and Angiogenesis
The evidence that microglia accounts for 30% of glioma mass has raised the question about its role in tumor growth and progression.
In 2002, an in vitro study evidenced that the motility of murine glioma cells, when in the presence of microglia, increased by three times [49], while endothelial cells and oligodendrocytes had little effect on glioma cells’ motility. In recent years, different soluble factors released by microglial cells have been identified as glioma growth and motility promoters [50]. TGF-β promotes glioma cells motility with processes that involve integrin and matrix-metalloproteinase induction [51]. In particular, TGF-β superfamily members 1–3 have been identified as immunosuppressive cytokines, which are upregulated in glioma tissues and secreted by tumoral cells [52]. Furthermore, through the activation of matrix-metalloproteinase (MMP)-2 and the suppression of tissue inhibitor of metalloproteinases (TIMP)-2, which degrades extracellular matrix, GAM-secreted TGF-β facilitates glioma cells motility in vitro [51]. To further confirm the importance of MMP-2 role in gliomas spread, a significant association between MMP-2 expression with astrocytomas aggressiveness and unfavorable prognosis has also been demonstrated [53].
To activate pro-MMP2, an inactive pro-form of MMP2, membrane-type1 matrix metalloproteinase 1 (MT1-MMP) is needed. Normal microglia do not normally elaborate MT1-MMP, while in glioma associated microglia MT1-MMP is overexpressed and its expression increases with glioma grade [54].
Toll-like receptor (TLR) 2 has been identified as an important MT1-MMP up-regulation promoter in microglia. There is an increasing list of TLRs endogenous ligand. For example, versican (or CSPG2) is a ECM component, member of chondroitin sulfate proteoglycans family, and it exists in four different iso-forms V0, V1, V2, and V3 (ref). V1 in the brain is responsible for neuronal differentiation and promotes neurite outgrowth, while V2 is an inhibitor of axonal growth [55][56]. In a study, Hu et al. demonstrated that versican V0/V1 was highly expressed by mouse and human gliomas compared with normal brain tissues. Moreover, versican activated macrophage through TLR2, while V1 iso-form significantly induced microglial MT1-MMP expression [37].
As further proof of TLR2 and MT1-MMP importance in glioma growth and progression, a study demonstrated that, in mice with deletion of TLR2, implanted gliomas were significantly smaller and mice survival prolonged compared with wild-type control mice. Furthermore, TLR2-neutralizing antibodies reduced microglial MT1-MMP expression and attenuated glioma growth [57].
The co-chaperone stress-inducible protein 1 (STI-1), a cellular prion protein ligand, synthesized and released by microglial cells, induces glioma cells migration and proliferation in vitro and in vivo [58].
As already mentioned before, CCL2 and its receptor CCR2, expressed on microglial cells surface, can induce IL-6 secretion and intensify glioma invasiveness [15].
Different studies demonstrated that microglia can induce angiogenesis by secreting several pro-angiogenic factors, such as vascular endothelial growth factor (VEGF) [59] and IL-6 [60]. (Figure 1) The administration of Sunitinib, a VEGFR inhibitor, combined with Bevacizumab, a VEGF inhibitor, reduced the infiltration of myeloid cells in gliomas and their vascularization [61]. IL-6 expression in microglial cells is mainly induced by the activation of the receptor for advanced glycation end products (RAGE) [62]. In a study, genetic depletion of RAGE in murine microglia abrogated angiogenesis by downregulating the expression of proangiogenic factors and was associated with animal prolonged survival [63].
All these evidences support the idea that genesis, growth, and progression of gliomas are not exclusively dependent on the intrinsic mutations of neoplastic cells, but are strongly influenced by the surrounding microenvironment and by the communication between tumoral and non-tumoral cells. Hence, it appears essential to strengthen the focus of scientific research on the study and the understanding of GAM in order to identify new therapeutic targets and treatment strategies for glioma patients, which are desperately needed.
References
- Fernandes, C. Current Standards of Care in Glioblastoma Therapy. In Glioblastoma; Codon Publications: Brisbane, Australia, 2017; pp. 197–241.
- Gómez-Oliva, R. Evolution of Experimental Models in the Study of Glioblastoma: Toward Finding Efficient Treatments. Front. Oncol. 2021, 10, 3245.
- Badie, B.; Schartner, J. Role of microglia in glioma biology. Microsc. Res. Technol. 2001, 54, 106–113.
- Graeber, M.B.; Scheithauer, B.W.; Kreutzberg, G.W. Microglia in brain tumors. Glia 2002, 40, 252–259.
- Azambuja, J.H.; Ludwig, N.; Yerneni, S.; Rao, A.; Braganhol, E.; Whiteside, T.L. Molecular profiles and immunomodulatory activities of glioblastoma-derived exosomes. Neuro-Oncol. Adv. 2020, 2, vdaa056.
- Najafi, M.; Farhood, B.; Mortezaee, K.; Kharazinejad, E.; Majidpoor, J.; Ahadi, R. Hypoxia in solid tumors: A key promoter of cancer stem cell (CSC) resistance. J. Cancer Res. Clin. Oncol. 2020, 146, 19–31.
- Sankowski, R. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat. Neurosci. 2019, 22, 2098–2110.
- Flgel, A.; Bradl, M.; Kreutzberg, G.W.; Graeber, M.B. Transformation of donor-derived bone marrow precursors into host microglia during autoimmune CNS inflammation and during the retrograde response to axotomy. J. Neurosci. Res. 2001, 66, 74–82.
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2015, 19, 20–27.
- Ajami, B.; Bennett, J.L.; Krieger, C.; McNagny, K.M.; Rossi, F.M.V. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat. Neurosci. 2011, 14, 1142–1149.
- Müller, A.; Brandenburg, S.; Turkowski, K.; Müller, S.; Vajkoczy, P. Resident microglia, and not peripheral macrophages, are the main source of brain tumor mononuclear cells. Int. J. Cancer 2015, 137, 278–288.
- Zhou, W. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 2015, 17, 170–182.
- Friebel, E. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 2020, 181, 1626–1642.e20.
- Platten, M. Monocyte chemoattractant protein-1 increases microglial infiltration and aggressiveness of gliomas. Ann. Neurol. 2003, 54, 388–392.
- Zhang, J.; Sarkar, S.; Cua, R.; Zhou, Y.; Hader, W.; Yong, V.W. A dialog between glioma and microglia that promotes tumor invasiveness through the CCL2/CCR2/interleukin-6 axis. Carcinogenesis 2012, 33, 312–319.
- Okada, M. Tumor-associated macrophage/microglia infiltration in human gliomas is correlated with MCP-3, but not MCP-1. Int. J. Oncol. 2009, 34, 1621–1627.
- Wang, S.C.; Hong, J.H.; Hsueh, C.; Chiang, C.S. Tumor-secreted SDF-1 promotes glioma invasiveness and TAM tropism toward hypoxia in a murine astrocytoma model. Lab. Investig. 2012, 92, 151–162.
- Ludwig, A.; Weber, C. Transmembrane chemokines: Versatile ‘special agents’ in vascular inflammation. Thromb. Haemost. 2007, 97, 694–703.
- Held-Feindt, J. CX3CR1 promotes recruitment of human glioma-infiltrating microglia/macrophages (GIMs). Exp. Cell Res. 2010, 316, 1553–1566.
- Feng, X. Loss of CX3CR1 increases accumulation of inflammatory monocytes and promotes gliomagenesis. Oncotarget 2015, 6, 15077–15094.
- Ku, M.C. GDNF mediates glioblastoma-induced microglia attraction but not astrogliosis. Acta Neuropathol. 2013, 125, 609–620.
- Coniglio, S.J. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol. Med. 2012, 18, 519–527.
- Pyonteck, S.M. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272.
- Sielska, M. Distinct roles of CSF family cytokines in macrophage infiltration and activation in glioma progression and injury response. J. Pathol. 2013, 230, 310–321.
- Casano, A.M.; Peri, F. Microglia: Multitasking specialists of the brain. Dev. Cell 2015, 32, 469–477.
- Bayerl, S.H. Time lapse in vivo microscopy reveals distinct dynamics of microglia-tumor environment interactions-a new role for the tumor perivascular space as highway for trafficking microglia. Glia 2016, 64, 1210–1226.
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665.
- Shaked, I.; Porat, Z.; Gersner, R.; Kipnis, J.; Schwartz, M. Early activation of microglia as antigen-presenting cells correlates with T cell-mediated protection and repair of the injured central nervous system. J. Neuroimmunol. 2004, 146, 84–93.
- Colton, C.A.; Wilcock, D.M. Assessing Activation States in Microglia. CNS Neurol. Disord. Drug Targets 2012, 9, 174–191.
- Boche, D.; Perry, V.H.; Nicoll, J.A.R. Review: Activation patterns of microglia and their identification in the human brain. Neuropathol. Appl. Neurobiol. 2013, 39, 3–18.
- Roberts, E.S.; Masliah, E.; Fox, H.S. CD163 identifies a unique population of ramified microglia in HIV encephalitis (HIVE). J. Neuropathol. Exp. Neurol. 2004, 63, 1255–1264.
- Zeiner, P.S. Distribution and prognostic impact of microglia/macrophage subpopulations in gliomas. Brain Pathol. 2019, 29, 513–529.
- Fabian, M.R.; Sonenberg, N. The mechanics of miRNA-mediated gene silencing: A look under the hood of miRISC. Nat. Struct. Mol. Biol. 2012, 19, 586–593.
- Roszer, T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediat. Inflamm. 2015, 2015, 816460.
- Watanabe, K.; Jose, P.J.; Rankin, S.M. Eotaxin-2 Generation Is Differentially Regulated by Lipopolysaccharide and IL-4 in Monocytes and Macrophages. J. Immunol. 2002, 168, 1911–1918.
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686.
- Hu, F. Glioma-derived versican promotes tumor expansion via glioma-associated microglial/macrophages Toll-like receptor 2 signaling. Neuro-Oncology 2015, 17, 200–210.
- Ummenthum, K. Pentraxin-3 is upregulated in the central nervous system during MS and EAE, but does not modulate experimental neurological disease. Eur. J. Immunol. 2016, 46, 701–711.
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J. Immunol. 2000, 164, 6166–6173.
- Komohara, Y.; Ohnishi, K.; Kuratsu, J.; Takeya, M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J. Pathol. 2008, 216, 15–24.
- Budhu, A. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell 2006, 10, 99–111.
- Gentles, A.J. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945.
- Hattermann, K. Chemokine expression profile of freshly isolated human glioblastoma- associated macrophages/microglia. Oncol. Rep. 2014, 32, 270–276.
- Zeiner, P.S. MIF Receptor CD74 is Restricted to Microglia/Macrophages, Associated with a M1-Polarized Immune Milieu and Prolonged Patient Survival in Gliomas. Brain Pathol. 2015, 25, 491–504.
- Gabrusiewicz, K.; Ellert-Miklaszewska, A.; Lipko, M.; Sielska, M.; Frankowska, M.; Kaminska, B. Characteristics of the alternative phenotype of microglia/macrophages and its modulation in experimental gliomas. PLoS ONE 2011, 6, e23902.
- Shemer, A.; Erny, D.; Jung, S.; Prinz, M. Microglia Plasticity during Health and Disease: An Immunological Perspective. Trends Immunol. 2015, 36, 614–624.
- Li, W.; Graeber, M.B. The molecular profile of microglia under the influence of glioma. Neuro-Oncology 2012, 14, 958–978.
- Lisi, L.; Laudati, E.; Navarra, P.; Russo, C.D. The mTOR kinase inhibitors polarize glioma-activated microglia to express a M1 phenotype. J. Neuroinflamm. 2014, 11, 125.
- Bettinger, I.; Thanos, S.; Paulus, W. Microglia promote glioma migration. Acta Neuropathol. 2002, 103, 351–355.
- Szulzewsky, F. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS ONE 2015, 10, e0116644.
- Wick, W.; Platten, M.; Weller, M. Glioma cell invasion: Regulation of metalloproteinase activity by TGF-β. J. Neurooncol. 2001, 53, 177–185.
- Kaminska, B.; Kocyk, M.; Kijewska, M. TGF beta signaling and its role in glioma pathogenesis. Adv. Exp. Med. Biol. 2013, 986, 171–187.
- Haresh, K. Granulocytic sarcoma masquerading as Ewing’s sarcoma: A diagnostic dilemma. J. Cancer Res. Ther. 2008, 4, 137–139.
- Markovic, D.S. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc. Natl. Acad. Sci. USA 2009, 106, 12530–12535.
- Wu, Y. Versican V1 Isoform Induces Neuronal Differentiation and Promotes Neurite Outgrowth. Mol. Biol. Cell 2004, 15, 2093–2104.
- Schmalfeldt, M.; Bandtlow, C.E.; Dours-Zimmermann, M.T.; Winterhalter, K.H.; Zimmermann, D.R. Brain derived versican V2 is a potent inhibitor of axonal growth. J. Cell Sci. 2000, 113, 807–816.
- Vinnakota, K. Toll-like receptor 2 mediates microglia/brain macrophage MT1-MMP expression and glioma expansion. Neuro-Oncology 2013, 15, 1457–1468.
- da Fonseca, A.C.C. Increased expression of stress inducible protein 1 in glioma-associated microglia/macrophages. J. Neuroimmunol. 2014, 274, 71–77.
- Brandenburg, S. Resident microglia rather than peripheral macrophages promote vascularization in brain tumors and are source of alternative pro-angiogenic factors. Acta Neuropathol. 2016, 131, 365–378.
- Piperi, C. Prognostic significance of IL-8-STAT-3 pathway in astrocytomas: Correlation with IL-6, VEGF and microvessel morphometry. Cytokine 2011, 55, 387–395.
- Piao, Y. Glioblastoma resistance to anti-VEGF therapy is associated with myeloid cell infiltration, stem cell accumulation, and a mesenchymal phenotype. Neuro-Oncology 2012, 14, 1379–1392.
- Kierdorf, K.; Fritz, G. RAGE regulation and signaling in inflammation and beyond. J. Leukoc. Biol. 2013, 94, 55–68.
- Chen, X. RAGE expression in tumor-associated macrophages promotes angiogenesis in glioma. Cancer Res. 2014, 74, 7285–7297.
More
Information
Subjects:
Neurosciences; Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
751
Revisions:
2 times
(View History)
Update Date:
20 Apr 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No