Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Urtė Andriušaitytė | -- | 2199 | 2022-04-14 21:38:30 | | | |
| 2 | Peter Tang | Meta information modification | 2199 | 2022-04-15 03:11:37 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Andriušaitytė, U.; Gulla, A.; , .; Strupas, K.; Kelly, H. Epithelial–Mesenchymal Transition and Metformin on Pancreatic Cancer Chemoresistance. Encyclopedia. Available online: https://encyclopedia.pub/entry/21799 (accessed on 10 August 2026).
Andriušaitytė U, Gulla A, , Strupas K, Kelly H. Epithelial–Mesenchymal Transition and Metformin on Pancreatic Cancer Chemoresistance. Encyclopedia. Available at: https://encyclopedia.pub/entry/21799. Accessed August 10, 2026.
Andriušaitytė, Urtė, Aiste Gulla, , Kęstutis Strupas, Helena Kelly. "Epithelial–Mesenchymal Transition and Metformin on Pancreatic Cancer Chemoresistance" Encyclopedia, https://encyclopedia.pub/entry/21799 (accessed August 10, 2026).
Andriušaitytė, U., Gulla, A., , ., Strupas, K., & Kelly, H. (2022, April 14). Epithelial–Mesenchymal Transition and Metformin on Pancreatic Cancer Chemoresistance. In Encyclopedia. https://encyclopedia.pub/entry/21799
Andriušaitytė, Urtė, et al. "Epithelial–Mesenchymal Transition and Metformin on Pancreatic Cancer Chemoresistance." Encyclopedia. Web. 14 April, 2022.
Copy Citation
Pancreatic cancer is among the most malignant and aggressive forms of neoplastic diseases, with pancreatic adenocarcinoma (PDAC) accounting for 90% of all pancreatic tumors. A particular feature of PDAC that has been identified, which significantly contributes to the dense tumor stroma and invasive potential, is the presence of epithelial–mesenchymal transition (EMT).
cancer stem cells
chemotherapy
epithelial–mesenchymal transition
metformin
pancreatic ductal carcinoma
1. Introduction
Pancreatic cancer is among the most malignant and aggressive forms of neoplastic diseases, with pancreatic adenocarcinoma (PDAC) accounting for 90% of all pancreatic tumors [1][2]. Although the 5-year survival rate has increased from 5% to 9% over the past decade, this still represents a dismal prognostic outcome [3][4]. The best chance of survival is for patients to be diagnosed at an early stage of PDAC, thus making them eligible for surgery; however, this represents only 20–30% of patients. Therefore, for many patients, surgery is not an option and current chemotherapy regimens, such as FOLFIRINOX and gemcitabine plus nab-paclitaxel, have limited effectiveness. As a result, there is a need to develop novel therapies, including immunotherapies and stroma-targeted methods [5]. Pancreatic tumors are dense, fibrotic masses that impede adequate drug delivery. The main histological hallmark of pancreatic cancer is the heterogeneous tumor microenvironment that dynamically evolves during neoplastic development due to an accumulation of mutations, which transforms normal mucosal cells to precursor lesions and finally to invasive malignancies [6][7]. Understanding the molecular pathways involved in the carcinogenesis, progression, metastasis, and tumor microenvironment growth are crucial for developing effective new therapies [7]. A particular feature of PDAC that has been identified, which significantly contributes to the dense tumor stroma and invasive potential, is the presence of epithelial–mesenchymal transition (EMT) [5]. Due to the abnormal pathological process of EMT, cancer cells obtain a mesenchymal phenotype and express specific extracellular matrix components, which substantially contributes to their migratory capacity. Moreover, various experiments elucidated that EMT gives rise to a specific pluripotent stem cell subpopulation, called cancer stem cells (CSCs), which have self-renewal properties and the ability to promote tumorigenicity [5]. As is explained further below, EMT also participates in specific cellular pathways related to the inhibition of autophagy and cell apoptosis, potentially causing epigenetic changes and a reduced expression of nucleoside transporters, thereby reducing the uptake of chemotherapeutic drugs. All of the above-mentioned factors affect drug transmission efficacy and greatly impact chemoresistance in PDAC. Consequently, research has increasingly focused on the necessity of a therapy addressing the dense stromal environment of the tumor, of which EMT is a contributing factor. Accumulating evidence revealed the potential of the anti-diabetic drug metformin to reverse EMT through diverse signaling pathways, thereby re-establishing chemosensitivity [8][9]. Metformin, through its ability to prevent the overexpression of EMT markers, such as ZEB, TWIST and SLUG (SNAIL2), is able to suppress the dynamic invasiveness and CSC formation in response to the neoplastic microenvironment. Furthermore, metformin inhibits EMT and PDAC metastatic potential through the downregulation of the mTOR pathway; hence, metformin’s ability to inhibit the EMT trans-differentiation process may represent a promising therapeutic strategy to clinically overcome chemotherapy refractoriness in CSC-enriched invasive pancreatic carcinomas [10].
2. Epithelial–Mesenchymal Transition
2.1. Epithelial–Mesenchymal Transition and Cancer Stem Cells
Epithelial cells in solid tumors are observed to undergo EMT, an abnormal pathological process in which cancer cells acquire a mesenchymal phenotype in response to signals they attain from the malignant microenvironment, particularly from reactive stromal cells [11]. Likewise, cells obtain migratory capacity, resistance to apoptosis and expression of extracellular matrix components [12]. These changes generate CSCs, a subset of undifferentiated neoplastic cells that can initiate heterogeneous cancers while triggering tumorigenesis. According to the “Seed and Soil” hypothesis, first proposed by Stephen Paget in 1889, cancer-initiating cells are endowed with higher tumor-forming potential and have the capacity to “reproduce” neoplasms when cells seed distant organs to develop metastases [13]. In PDAC, cancer initiating cells have been specified as those expressing CD44, CD24 and ESA, forming tumors at a higher frequency, compared to cells that do not express this phenotype [14]. In recent studies, it was demonstrated that more than 65% of PDAC patient-derived organoids had characteristics of CSC, characterized as CD44+ CD24+ [15]. The EMT phenomenon has been directly related to cancer stem cell features, since CSCs also express an EMT phenotype in various types of cancer, including PDAC. In an analysis of breast cancer, it was observed that the upregulation of EMT transcriptional factors, such as TWIST or SNAI, made the cells more mesenchymal and elevated the expression of CD44 and CD24 [15]. It was also proposed that the EMT regulator ZEB1 enforces isoforms of CD44, while CD44s activates the expression of ZEB1. As a result, a self-sustaining expression of ZEB1 and CD44 is achieved, thereby linking EMT with the stem cell properties in PDAC [16]. Therefore, it can be considered that EMT contributes to pancreatic cancer stem cells’ migratory capacity by maintaining their ability to multiply and participate in the production of progenies in metastasis. Apart from CD44+ CD24+ ESA+ populations of pancreatic cancer stem cells, c-Met (mesenchymal–epithelial transition factor) has also been identified as a potential prognostic factor for PDAC [17]. c-Met is a receptor tyrosine kinase found on the surface of epithelial cells. In normal circumstances, c-Met and its ligand HGF/SF (hepatocyte growth factor/scatter factor) moderate tissue regeneration, wound healing and the formation of nerves and muscles [18]. The abnormal activation of c-Met can promote the development and progression of multiple cancers [19]. For instance, a recent analysis compared the median survival for PDAC patients, revealing 21.65 months of viability for negative c-Met patients and only 9.45 months for positive ones [20]. In this regard, high c-Met expression is closely associated with poor prognosis in PDAC patients. Several other processes that occur during EMT include activation of transcription factors, such as the key regulators, SNAI1, SNAI2, ZEB, and TWIST factors [21]. These EMT regulators have been shown to repress the expression of E-cadherin, a key epithelial marker. The downregulation of E-cadherin impedes cellular adhesion and imparts cellular motility, which is correlated with reduced chemotherapeutic drug sensitivity [22][23]. Concurrently, the activation of snail proteins upregulates the expression of mesenchymal proteins, such as vimentin [24]. Vimentin is usually found as the main intermediate filament protein of normal mesenchymal tissue. Vimentin functions to maintain cellular integrity and provides resistance against stress factors, and its expression has been detected in epithelial malignancies, such as gastrointestinal tumors. During EMT, pancreatic cells change expression from keratin- to vimentin-type intermediate filaments and become resistant to programed cell death [25] (Figure 1).
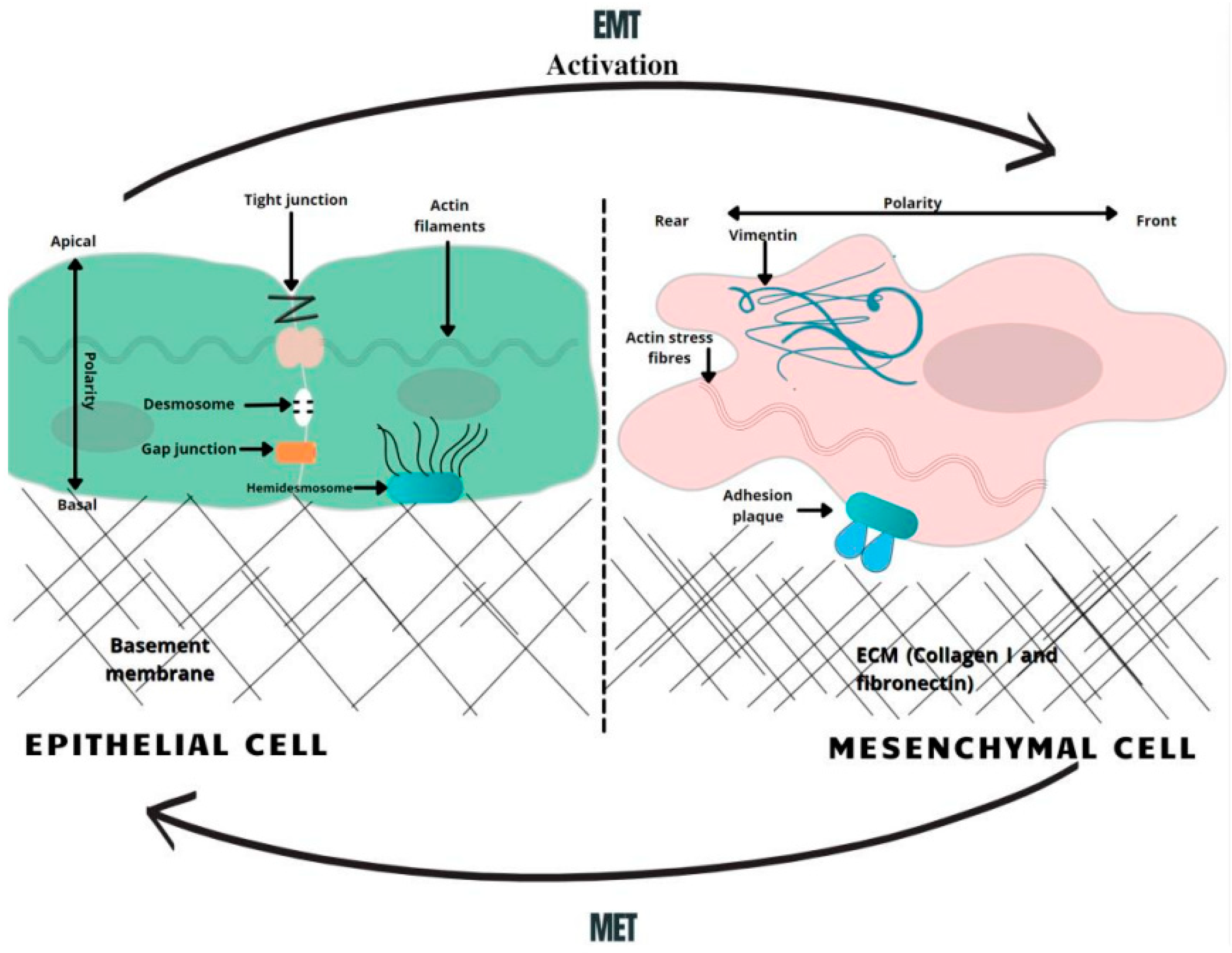
Figure 1. A schematic overview of EMT-related changes in the cell morphology. In the course of EMT, cells lose their normal tight, adherent and gap junctions, retaining only minimal cellular connections. Correspondingly, the adhesion belt made of actin filaments is changed to loose actin stress fibers. Therefore, the activation of the EMT generates profound modulations in cell physiology, especially affecting cell–cell junctions, cytoskeletal arrangement, cell–cell interactions, and the composition of the extracellular matrix (ECM), as well as completely changing cell polarity [26]. MET—mesenchymal-to-epithelial transition, EMT—epithelial-to-mesenchymal transition.
2.2. Chemoresistance and EMT Impact on Oncotherapy
Taking into consideration all of the above-mentioned mechanisms, it is now widely accepted that the EMT-associated modifications of gene expression and CSC populations both have crucial roles in making cells resistant to current clinical therapies [26][27]. Dynamic and proliferative CSC populations with high cellular plasticity, including a unique self-renewal capacity, which is critical for metastasis and chemoresistance, subsequently makes them resistant to conventional chemotherapy; hence, CSCs are a crucial target when considering novel treatments [28]. Moreover, traditional chemotherapy targets proliferating cells, while CSCs have been widely described as being slow-cycling and quiescent, which is a prominent mechanism for resistance to the conventional therapies [29]. Hence, CSC-targeted chemotherapeutic regimens have the potential to suppress tumor progression, inhibit their metastasis-forming ability and ultimately overcome therapeutic resistance, including relapses, which are frequently observed in response to current treatment methods [30]. Studies with gemcitabine, 5-FU (5-fluorouracil) and cisplatin have shown that EMT-type cells are resistant to chemotherapy, while non-EMT-type cells are susceptible to these chemotherapeutic drugs. Altogether, EMT plays an important role in the regulation of treatment resistance in PDAC, suggesting that targeting EMT could not only reduce and suppress the population of CSCs that have been implicated in tumor metastasis, but also contribute to an increased sensitivity to chemotherapy [31].
Collectively, EMT is involved in mediating NF-kB and AKT activity, elements that are known to be major regulators of autophagy processes, with both leading to cell apoptosis [23]. EMT influences neoplastic cells to gain a CSC phenotype and enhances their neoplastic replicative capacity, as well as increasing chemotherapeutic resistance [32][33]. Other factors determining EMT-initiated chemoresistance include epigenetic changes, which reduce the expression of the hENT1 nucleoside transporters needed for gemcitabine uptake. CSCs showing an EMT phenotype are prone to express ATP-binding cassette (ABC) transporters [34]. The overexpression of ABC family efflux transporters also increases the efflux of therapeutic agents from cells, e.g., the MDR1 and MRP1 ABC efflux transporters initiate a resistance to nucleoside analogues, such as gemcitabine [35]. EMT also initiates the activation of inert fibroblasts in the tumor microenvironment by modifying epithelial cells to active myofibroblasts. Fibro-inflammatory stroma (the desmoplastic stroma) is made of dense ECM deposited mostly by cancer-associated fibroblasts [36][37]. A dense stroma surrounds the tumor and increases the tumor interstitial fluid pressure, leading to poor tumor elasticity and the decreased perfusion and efficacy of administered therapy [33].
Furthermore, EMT activates specific mechanisms that help cancer cells avoid the effect of cytotoxic T cells. These include increased expression of PD-L1, which binds to inhibitory immune-checkpoint receptors, expressed by T cells, thereby diminishing their function. The activity of T cells is also inhibited by the elevated secretion of thrombospondin-1, which normally stimulates the maturation of T cells in the tumor microenvironment. The inhibition of thrombospondin-1 consequently disrupts the activity and development of T cells [26].
EMT therefore plays a critical role in inducing stem cell features, which have been linked to enhanced invasiveness and aggressiveness, ensuring that cell dissemination from the primary tumor location to the metastasis site occurs even before pancreatic cancer lesions are detected [38]. This phenomenon helps to explain the parallel low survival rate of PDAC patients with increased EMT, whereby they have an impaired capacity to respond to current chemotherapeutic methods [39] (Figure 2).
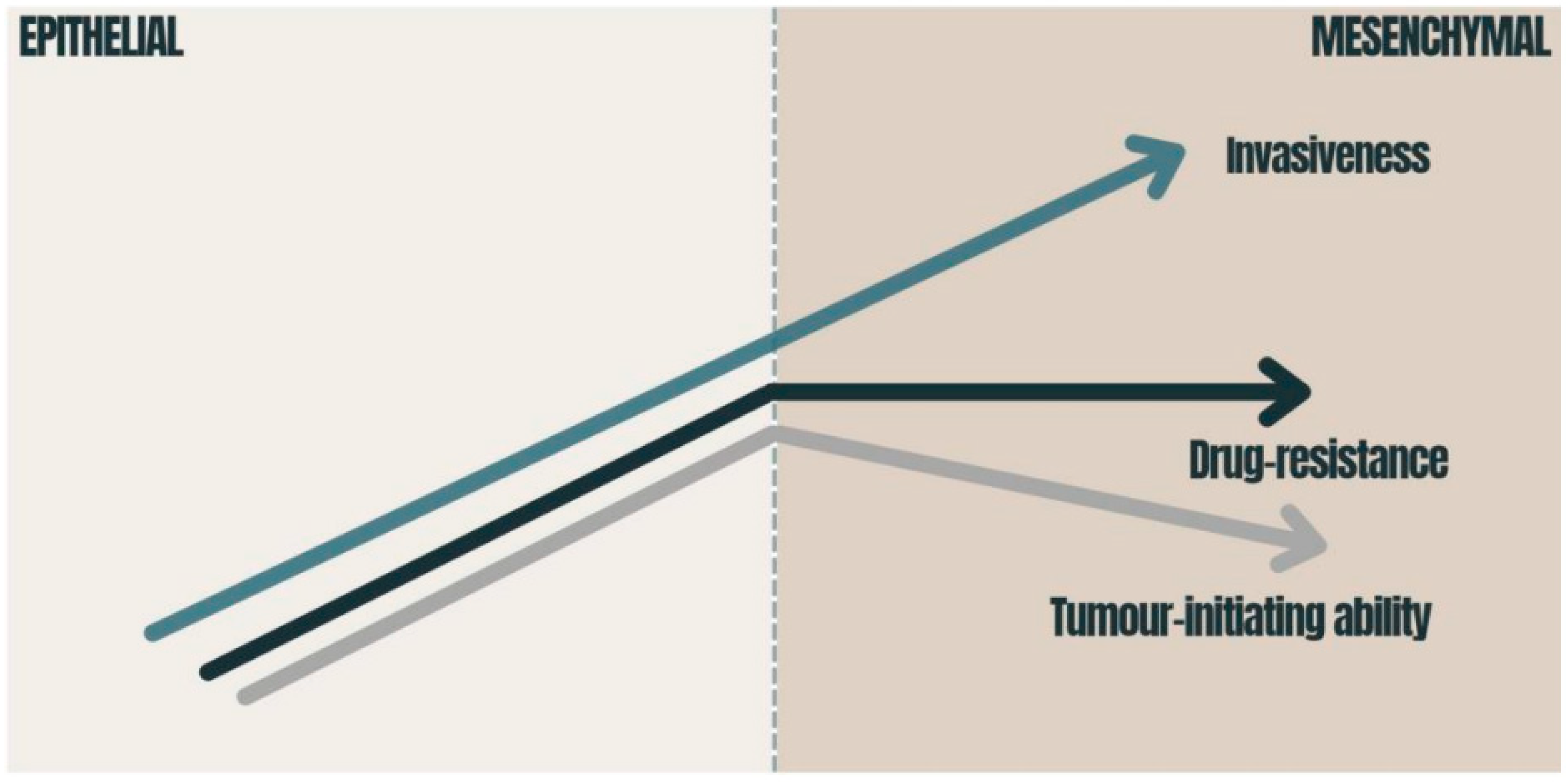
Figure 2. Consequences of EMT in carcinomas: the graphic presents how the tumor-initiating ability, invasiveness and degree of chemoresistance change during EMT activation. The tumor-initiating ability is affected by the degree of EMT activation; immense EMT activation has a deleterious effect on the tumor-initiating ability [40]. Drug resistance is also related to EMT and reaches its maximum at an intermediate level of EMT activation [40][41]. The migration of cancer cells requires the strong activation of the EMT program [42]. EMT—epithelial-to-mesenchymal transition.
3. Metformin and PDAC
Metformin is the most widely used oral anti-hyperglycemic agent in the treatment of type 2 diabetes, where it acts to lower glucose through enhancing the activity of insulin [43][44]. However, a number of epidemiological studies identified an association of improved survival outcomes among PDAC patients being treated with metformin in combination therapy (compared to those receiving a monotherapy) [45][46]. With a random model approach, there are important differences in the overall survival (HR = 0.85, 95% CI: 0.77–0.94, p = 0.002) between PDAC patients who were treated with metformin and underwent pancreatectomy and those who underwent pancreatectomy without metformin treatment [47]. A novel retrospective study of diabetic patients with pancreatic cancer also found that the mean overall survival time was 15.2 months for the metformin-treated group and 11.1 months for the control group. These results were only statistically significant in patients with non-metastatic lesions [48]. Thus, the correlation between metformin usage and its role in beneficial outcomes on PDAC treatment is now a focus of ongoing research to identify effective novel therapeutic strategies for PDAC.
Metformin has been identified as having the potential to modulate multiple molecular pathways crucial to malignant cell progression and metabolism by inhibiting features of aggressive tumors, such as desmoplasia and EMT reactions. The desmoplastic reaction, resulting from the proliferation of activated pancreatic stellate cells (PSCs) and the increased deposition of extracellular matrix components, is a significant pathological characteristic of pancreatic neoplasms, while EMT is responsible for invasion-metastasis cascades in pancreatic cancer cells [49]. As TGF-β1 is a potent inducer of EMT, accumulating evidence suggests that an inhibition of TGF-β1 production in PDAC cells by metformin-induced AMPK activation inhibits malignant pancreatic cell growth [50].
The novel advances elucidating the molecular mechanisms that are relevant for metformin usage in PDAC treatment demonstrated that it can induce the apoptosis by augmenting the activity caspases, such as caspase-3 and -8. Caspase-8, as a highly promising putative target, leads to the activation of caspase-3, -6, and -7, which directly induce the cell-death pathway [51]. Furthermore, metformin inhibits neoplastic angiogenesis via suppressing the HIF-1α-induced expression of angiogenesis-associated factors (AAFs) and downregulating the expression of the vascular endothelial growth factor (VEGF) [52]. By the same token, metformin demonstrates a great promise in improving current cancer therapies, as it inhibits the matrix metalloproteinases ((MMP)-2 and (MMP)-9) and suppresses the migration of tumorigenic endothelial cells. One of the most important factors in the metastasis of PDAC cells is matrix metalloproteinase MMPs, which destroys the extracellular matrix and allows malignant pancreatic cells to form metastasis. These anticancer properties appear to synergize with already existing chemotherapeutics, which allow us to postulate that metformin might play a role in PDAC prevention and therapeutic treatment, whereas it counteracts malignant cell development and growth on specific levels [51][53].
References
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620.
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27.
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424.
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res. 2014, 74, 2913–2921.
- Chandana, S.R.; Babiker, H.M.; Mahadevan, D. Therapeutic trends in pancreatic ductal adenocarcinoma (PDAC). Expert Opin. Investig. Drugs 2019, 28, 161–177.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Hessmann, E.; Buchholz, S.M.; Demir, I.E.; Singh, S.K.; Gress, T.M.; Ellenrieder, V.; Neesse, A. Microenvironmental determinants of pancreatic cancer. Physiol. Rev. 2020, 100, 1707–1751.
- Candido, S.; Abrams, S.L.; Steelman, L.; Lertpiriyapong, K.; Martelli, A.M.; Cocco, L.; Ratti, S.; Follo, M.Y.; Murata, R.M.; Rosalen, P.L.; et al. Metformin influences drug sensitivity in pancreatic cancer cells. Adv. Biol. Regul. 2018, 68, 13–30.
- Chen, K.; Qian, W.; Jiang, Z.; Cheng, L.; Li, J.; Sun, L.; Zhou, C.C.; Gao, L.P.; Lei, M.; Yan, B.; et al. Metformin suppresses cancer initiation and progression in genetic mouse models of pancreatic cancer. Mol. Cancer 2017, 16, 131.
- Del Barco, S.; Vazquez-Martin, A.; Cufí, S.; Oliveras-Ferraros, C.; Bosch-Barrera, J.; Joven, J.; Martin-Castillo, B.; Menendez, J.A. Metformin: Multi-faceted protection against cancer. Oncotarget 2011, 2, 896–917.
- Zhou, P.T.; Li, B.; Liu, F.R.; Zhang, M.C.; Wang, Q.; Liu, Y.H.; Yao, Y.; Li, D. The epithelial to mesenchymal transition (EMT) and cancer stem cells: Implication for treatment resistance in pancreatic cancer. Mol. Cancer 2017, 16, 52.
- Beuran, M.; Negoi, I.; Paun, S.; Ion, A.D.; Bleotu, C.; Negoi, R.I.; Hostiuc, S. The epithelial to mesenchymal transition in pancreatic cancer: A systematic review. Pancreatology 2015, 15, 217–225.
- Lehúede, C.; Dupuy, F.; Rabinovitch, R.; Jones, R.G.; Siegel, P.M. Metabolic plasticity as a determinant of tumor growth and metastasis. Cancer Res. 2016, 76, 5201–5208.
- Gzil, A.; Zarębska, I.; Bursiewicz, W.; Antosik, P.; Grzanka, D.; Szylberg, Ł. Markers of pancreatic cancer stem cells and their clinical and therapeutic implications. Mol. Biol. Rep. 2019, 46, 6629–6645.
- Choi, J.-I.; Jang, S.I.; Hong, J.; Kim, C.H.; Kwon, S.S.; Park, J.S.; Lim, J.-B. Cancer-initiating cells in human pancreatic cancer organoids are maintained by interactions with endothelial cells. Cancer Lett. 2021, 498, 42–53.
- Preca, B.-T.; Bajdak, K.; Mock, K.; Sundararajan, V.; Pfannstiel, J.; Maurer, J.; Wellner, U.; Hopt, U.T.; Brummer, T.; Brabletz, S.; et al. A self-enforcing CD44s/ZEB1 feedback loop maintains EMT and stemness properties in cancer cells. Int. J. Cancer 2015, 137, 2566–2577.
- Li, C.; Wu, J.; Hynes, M.; Dosch, J.; Sarkar, B.; Welling, T.H.; di Magliano, M.P.; Simeone, D.M. c-Met is a marker of pancreatic cancer stem cells and therapeutic target. Gastroenterology 2011, 141, 2218–2227.e5.
- Zhang, Y.Z.; Xia, M.F.; Jin, K.; Wang, S.F.; Wei, H.; Fan, C.M.; Wu, Y.F.; Li, X.L.; Li, X.Y.; Li, G.Y.; et al. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol. Cancer 2018, 17, 20.
- Neuzillet, C.; Couvelard, A.; Tijeras-Raballand, A.; de Mestier, L.; de Gramont, A.; Bédossa, P.; Paradis, V.; Sauvanet, A.; Bachet, J.-B.; Ruszniewski, P.; et al. High c-Met expression in stage I–II pancreatic adenocarcinoma: Proposal for an immunostaining scoring method and correlation with poor prognosis. Histopathology 2015, 67, 664–676.
- Lux, A.; Kahlert, C.; Grützmann, R.; Pilarsky, C. c-Met and PD-l1 on circulating exosomes as diagnostic and prognostic markers for pancreatic cancer. Int. J. Mol. Sci. 2019, 20, 3305.
- Jiang, J.H.; Liu, C.; Cheng, H.; Lu, Y.; Qin, Y.; Xu, Y.F.; Xu, J.; Long, J.; Liu, L.; Ni, Q.X.; et al. Epithelial-mesenchymal transition in pancreatic cancer: Is it a clinically significant factor? Biochim. Biophys. Acta-Rev. Cancer 2015, 1855, 43–49.
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196.
- Schober, M.; Jesenofsky, R.; Faissner, R.; Weidenauer, C.; Hagmann, W.; Michl, P.; Heuchel, R.L.; Haas, S.L.; Löhr, J.-M. Desmoplasia and chemoresistance in pancreatic cancer. Cancers 2014, 6, 2137–2154.
- Gradiz, R.; Silva, H.C.; Carvalho, L.; Botelho, M.F.; Mota-Pinto, A. MIA PaCa-2 and PANC-1—pancreas ductal adenocarcinoma cell lines with neuroendocrine differentiation and somatostatin receptors. Sci. Rep. 2016, 6, 21648.
- Wu, S.; Du, Y.; Beckford, J.; Alachkar, H. Upregulation of the EMT marker vimentin is associated with poor clinical outcome in acute myeloid leukemia. J. Transl. Med. 2018, 16, 170.
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629.
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726.
- Sancho, P.; Alcala, S.; Usachov, V.; Hermann, P.C.; Sainz, B. The ever-changing landscape of pancreatic cancer stem cells. Pancreatology 2016, 16, 489–496.
- Lyle, S.; Moore, N. Quiescent, slow-cycling stem cell populations in cancer: A review of the evidence and discussion of significance. J. Oncol. 2010, 2011, 396076.
- Bhagwandin, V.J.; Bishop, J.M.; Wright, W.E.; Shay, J.W. The Metastatic Potential and Chemoresistance of Human Pancreatic Cancer Stem Cells. PLoS ONE 2016, 11, e0148807.
- Hu, X.; Chen, W. Role of epithelial-mesenchymal transition in chemoresistance in pancreatic ductal adenocarcinoma. World J. Clin. Cases 2021, 9, 4998.
- Celià-Terrassa, T.; Jolly, M.K. Cancer stem cells and epithelial-to-mesenchymal transition in cancer metastasis. Cold Spring Harb. Perspect. Med. 2020, 10, a036905.
- Cannon, A.; Thompson, C.; Hall, B.R.; Jain, M.; Kumar, S.; Batra, S.K. Desmoplasia in pancreatic ductal adenocarcinoma: Insight into pathological function and therapeutic potential. Genes Cancer 2018, 9, 78–86.
- Li, Y.; Kong, D.; Ahmad, A.; Bao, B.; Sarkar, F.H. Pancreatic cancer stem cells: Emerging target for designing novel therapy. Cancer Lett. 2013, 338, 94–100.
- Adamska, A.; Falasca, M. ATP-binding cassette transporters in progression and clinical outcome of pancreatic cancer: What is the way forward? World J. Gastroenterol. 2018, 24, 3222–3236.
- Awaji, M.; Singh, R.K. Cancer-associated fibroblasts’ functional heterogeneity in pancreatic ductal adenocarcinoma. Cancers 2019, 11, 290.
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596.
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361.
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695.e4.
- Bierie, B.; Pierce, S.E.; Kroeger, C.; Stover, D.G.; Pattabiraman, D.R.; Thiru, P.; Donaher, J.L.; Reinhardt, F.; Chaffer, C.L.; Keckesova, Z.; et al. Integrin-β4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2337–E2346.
- Pattabiraman, D.R.; Bierie, B.; Kober, K.I.; Thiru, P.; Krall, J.A.; Zill, C.; Reinhardt, F.; Tam, W.L.; Weinberg, R.A. Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science 2016, 351, aad3680.
- Clark, A.G.; Vignjevic, D.M. Modes of cancer cell invasion and the role of the microenvironment. Curr. Opin. Cell Biol. 2015, 36, 13–22.
- Schernthaner, G.; Schernthaner, G.H. The right place for metformin today. Diabetes Res. Clin. Pract. 2020, 159, 107946.
- Gong, L.; Goswami, S.; Giacomini, K.M.; Altman, R.B.; Klein, T.E. Metformin pathways: Pharmacokinetics and pharmacodynamics. Pharm. Genom. 2012, 22, 820–827.
- Lee, M.-S.; Hsu, C.-C.; Wahlqvist, M.L.; Tsai, H.-N.; Chang, Y.-H.; Huang, Y.-C. Type 2 diabetes increases and metformin reduces total, colorectal, liver and pancreatic cancer incidences in Taiwanese: A representative population prospective cohort study of 800,000 individuals. BMC Cancer 2011, 11, 20.
- Coyle, C.; Cafferty, F.H.; Vale, C.; Langley, R.E. Metformin as an adjuvant treatment for cancer: A systematic review and meta-analysis. Ann. Oncol. 2016, 27, 2184–2195.
- Wan, G.; Sun, X.; Li, F.; Wang, X.; Li, C.; Li, H.; Yu, X.; Cao, F. Cellular Physiology and Biochemistry Cellular Physiology and Biochemistry Survival Benefit of Metformin Adjuvant Treatment For Pancreatic Cancer Patients: A Systematic Review and Meta-Analysis. Cell Physiol. Biochem. 2018, 49, 837–847.
- Sadeghi, N.; Abbruzzese, J.L.; Yeung, S.C.J.; Hassan, M.; Li, D. Metformin use is associated with better survival of diabetic patients with pancreatic cancer. Clin. Cancer Res. 2012, 18, 2905–2912.
- Duan, W.X.; Chen, K.; Jiang, Z.D.; Chen, X.; Sun, L.K.; Li, J.H.; Lei, J.J.; Xu, Q.H.; Ma, J.G.; Li, X.Q.; et al. Desmoplasia suppression by metformin-mediated AMPK activation inhibits pancreatic cancer progression. Cancer Lett. 2017, 385, 225–233.
- Lamouille, S.; Connolly, E.; Smyth, J.W.; Akhurst, R.J.; Derynck, R. TGf-β-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. Development 2012, 139, 1259–1273.
- Khezri, M.R.; Melekinejad, H.; Majidi-Zolbanin, N.; Ghasemnejad-Berenji, M. Anticancer potential of metformin: Focusing on gastrointestinal cancers. Cancer Chemother. Pharmacol. 2021, 87, 587–598.
- Ma, R.; Yi, B.; Riker, A.I.; Xi, Y. Metformin and cancer immunity. Acta Pharmacol. Sin. 2020, 41, 1403–1409.
- Gyawali, M.; Venkatesan, N.; Ogeyingbo, O.D.; Bhandari, R.; Botleroo, R.A.; Kareem, R.; Ahmed, R.; Elshaikh, A.O. Magic of a Common Sugar Pill in Cancer: Can Metformin Raise Survival in Pancreatic Cancer Patients? Cureus 2021, 13, e16916.
More
Information
Subjects:
Medicine, Research & Experimental
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
581
Revisions:
2 times
(View History)
Update Date:
15 Apr 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No