Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ahmad Najem | -- | 3396 | 2022-04-08 10:44:33 | | | |
| 2 | Beatrix Zheng | + 3 word(s) | 3399 | 2022-04-08 11:39:38 | | | | |
| 3 | Beatrix Zheng | Meta information modification | 3399 | 2022-04-08 11:40:37 | | | | |
| 4 | Beatrix Zheng | Meta information modification | 3399 | 2022-04-08 11:41:55 | | | | |
| 5 | Beatrix Zheng | + 1 word(s) | 3400 | 2022-04-11 08:53:28 | | | | |
| 6 | Beatrix Zheng | + 2 word(s) | 3402 | 2022-04-13 12:06:46 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Najem, A.; Ghanem, G.E. New Vulnerabilities of Melanoma. Encyclopedia. Available online: https://encyclopedia.pub/entry/21506 (accessed on 30 June 2026).
Najem A, Ghanem GE. New Vulnerabilities of Melanoma. Encyclopedia. Available at: https://encyclopedia.pub/entry/21506. Accessed June 30, 2026.
Najem, Ahmad, Ghanem E. Ghanem. "New Vulnerabilities of Melanoma" Encyclopedia, https://encyclopedia.pub/entry/21506 (accessed June 30, 2026).
Najem, A., & Ghanem, G.E. (2022, April 08). New Vulnerabilities of Melanoma. In Encyclopedia. https://encyclopedia.pub/entry/21506
Najem, Ahmad and Ghanem E. Ghanem. "New Vulnerabilities of Melanoma." Encyclopedia. Web. 08 April, 2022.
Copy Citation
Melanoma cells are notorious for their high plasticity and ability to switch back and forth between various melanoma cell states, enabling the adaptation to sub-optimal conditions and therapeutics. This phenotypic plasticity, which has gained more attention in cancer research, is proposed as a new paradigm for melanoma progression. Here the researchers present several rational therapeutic approaches, such as exploiting phenotype-specific and metabolic vulnerabilities and targeting components and signals of the tumor microenvironment (TME), to improve the response of melanoma patients to treatments.
melanoma
phenotype switching
tumor microenvironment
oxidative stress
metabolic reprogramming
therapeutic strategies
1. Introduction
Metastatic melanoma is notoriously one of the most difficult cancers to treat. The MAPK pathway is constitutively activated in the vast majority (90%) of cutaneous melanoma. BRAF (50%) and NRAS (20–30%) are the most frequent mutations followed by NF1 (10–14%) and KIT (5–10%). These genomic alterations are considered driver mutations in melanoma development [1][2]. MAPK inhibitors and immune checkpoint inhibitors have revolutionized the treatment of metastatic melanoma. Despite these recent advances, many patients do not respond to these therapies, and the acquired resistance remains a major problem [1][2].
The acquired resistance can be driven by genetic alterations activating the MAPK pathway, such as BRAF amplification, expression of BRAF splicing variants, NRAS and MEK mutations, or by MAPK-independent alterations, such as the activation of the PI3K/AKT pathway (PIK3CA mutations, AKT mutations/amplifications, and PTEN loss), and cyclin D1 amplifications [1][2]. The intertumoral and intratumoral genetic heterogeneities are also major obstacles to targeted therapy. Indeed, the emergence of distinct subclones with various mutations, including drug-resistant ones within the tumor or among different tumor sites, can lead to disease relapse [3].
However, melanoma cells are not only able to adapt to therapies by acquiring genetic alterations but they can also switch their cellular phenotype in order to adapt to therapeutics and various stressful conditions. This phenotypic plasticity, which has gained more attention in cancer research, is proposed as a new paradigm for melanoma progression and resistance to therapy. Such plasticity, referred to as phenotype switching in melanoma, is linked to epithelial–mesenchymal transition (EMT) and is characterized as dynamic cell state transitions that involve reversible transcriptional changes and epigenetic modifications in contrast to the irreversible genetic alterations [4][5][6][7]. Phenotypic plasticity is an important source of tumor heterogeneity and presents a major challenge for both targeted and immunotherapies.
The phenotype switching model in melanoma was initially described by Hoek et al. (2006) based on gene expression profiling data from both cell lines and melanoma patient samples [5][7]. This model is controlled by MITF a master lineage transcription factor and EMT transcription factors (TFs). According to this model, melanoma cells can interconvert between two main phenotypes, the melanocytic (differentiated/proliferative) state and the mesenchymal-like (invasive/undifferentiated) state [5][7].
2. Targeting Phenotypic Plasticity: Identifying New Vulnerabilities of Melanoma Phenotypes and Future Challenges
A deep understanding of the distinct melanoma phenotypes, their key regulators, the associated metabolic pathways, and the microenvironment signals is critical to identify and explore new vulnerabilities, which can constitute a very attractive strategy to overcome therapy resistance (Figure 1 and Table 1).
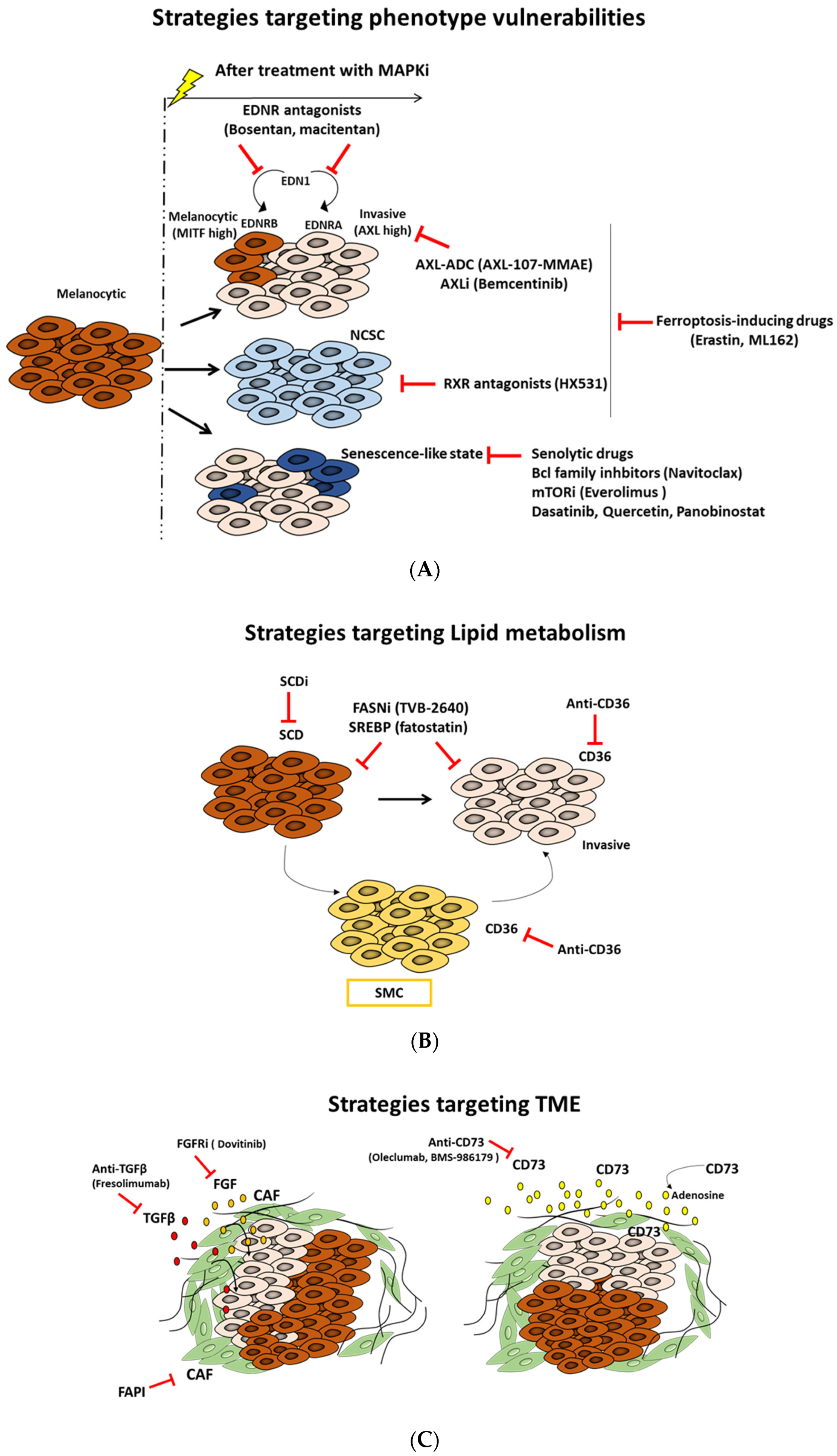
Figure 1. Targeted therapeutic approaches based on melanoma states vulnerabilities, metabolic rewiring, and microenvironment modulation. Schematic description of different therapeutic strategies: (A) exploiting vulnerabilities of distinct phenotypes, (B) targeting lipid metabolism, and (C) modulating components and signals of tumor microenvironment (TME).
Table 1. Targeting phenotypic plasticity: an overview of innovative and rational approaches for melanoma treatment.
| Therapeutic Approach | Targets/Drugs | Stage of Development | References |
|---|---|---|---|
| Targeting phenotype-specific vulnerabilities | Invasive phenotype: AXL-(ADC): AXL-107-MMAE Enapotamab vedotin |
Solid tumor phases I, II NCT02988817 |
[8][9] |
| Invasive phenotype: Small molecule AXLi (bemcentinib, dubermatinib) |
Solid tumor phases I, II Bemcentinib NCT03184571; NCT03649321 Dubermatinib: NCT02729298 |
[10][11] | |
| Invasive phenotype: Multi-targeted TKI (sitravatinib) |
Solid tumor phases I, II, III Sitravatinib NCT04518046; NCT03666143 NCT04123704;NCT03906071 |
[12] | |
| Neural crest stem cell (NCSC) state: pan-RXR antagonist (HX531) |
Preclinical | [13] | |
| Invasive/NCSC phenotypes: ferroptosis inducing drugs (erastin, ML162, and ML21s) |
Preclinical | [14] | |
| Phenotypic heterogeneity/TME: EDNR antagonists |
Bosentan NCT04158635 | [15] | |
| Senescence-like phenotype: Senolytics AKT/mTORi HDACi Bcl family inhibitor |
Solid tumors phases I, II, III mTORi: everolimus (NCT00876395) HDACi: panobinostat (NCT04897880) Navitoclax (NCT03366103) |
[16][17] | |
| Targeting lipid metabolism | Lipid uptake: Anti-CD36 | Preclinical | [18] |
| Lipid synthesis and uptake: SREBPi: fatostatin, botulin, and PF-429242 | Preclinical | [19] | |
| Lipogenesis: FASNi (TVB-2640) |
Solid tumor phase II NCT03179904; NCT03808558 |
[20][21] | |
| Targeting glutamine metabolism | Inhibition of GLS1: CB-839 | Phase I/II evaluation of CB-839 in combination with nivolumab in melanoma patients (NCT02771626) | [22][23] |
| Targeting TME components and signals | FAPI | Preclinical | [24][25] |
| Anti-TGF-beta | Solid tumor phase I NCT00356460 |
[26] | |
| Small molecule FGFRi | Dovitinib NCT01831726 NCT01676714 |
[27] | |
| Anti-CD73 | Solid tumor phases I, II Oleclumab: NCT03611556; NCT03381274; NCT04668300 CPI-006: NCT03454451 BMS-986179: NCT02754141 |
[28][29][30] |
ADC: antibody-drug conjugate; RXR: retinoid X receptor; TME: tumor microenvironment; FASNi: fatty acid synthase inhibitors, SREBPi: sterol regulatory element-binding proteins inhibitors; FAPI: fibroblast activation protein inhibitors.
2.1. Targeting Phenotype-Specific Vulnerabilities
2.1.1. Targeting AXL Receptor Tyrosine Kinase as a New Promising Approach
Axl is a member of the Tyro3, Axl, Mer (TAM) subfamily of receptor tyrosine kinases (RTKs). AXL is overexpressed in several cancer types and correlates with poor survival. AXL is activated by GAS6 ligand and can promote cell invasion, EMT, and angiogenesis. Moreover, AXL regulates the immune response and is implicated in the stem cell maintenance. More important, AXL contributes to drug resistance in cancer treatment [9].
In melanoma, AXL expression is associated with an invasive phenotype and thereby resistance to MAPKi. Thus, it represents an important target to inhibit this phenotype. Accordingly, it was demonstrated that the combination of AXL-107-MMAE with MAPKi cooperate to inhibit melanoma growth in vitro and in vivo [8]. AXL-107-MMAE is a specific AXL-targeted antibody–drug conjugate (ADC) generated by conjugating the human antibody with the microtubule disrupting agent monomethyl auristatin E (MMAE). AXL-107-MMAE is safe and tolerable and its efficacy was investigated in several tumor types [8]. Recently, Boshuizen al. 2021 showed that targeting immunotherapy-resistant melanoma and lung cancer using an AXL-targeting ADC enhances the sensitivity to immune checkpoint inhibitors (IC) [31].
Small molecule AXL inhibitors have also been proposed as new therapeutic strategies in melanoma. Indeed, bemcentinib is an oral, highly selective AXL inhibitor that shows promising results in preclinical and clinical studies, both alone and in combination, mainly with immune checkpoint inhibitors in multiple solid and hematologic tumors [10][11]. For instance, bemcentinib was investigated in combination with either dabrafenib/trametinib (D/T) or IC pembrolizumab in melanoma patients [11]. Dubermatinib is another selective AXL inhibitor that was investigated in CLL (NCT03572634) and solid tumors (NCT02729298). Other non-selective AXL inhibitors that have the latter among their targets, such as crizotinib, bosutinib, and cabozantinib, and multi-targeted TKI, such as sunitinib, foretinib, and sitravatinib, are also available in clinics [12].
Altogether, these findings show that targeting AXL can be a central approach to suppress the invasive phenotype in melanoma.
2.1.2. Targeting The Neural Crest Stem Cell (NCSC) State and RXRG Signaling to Delay Melanoma Relapse
In melanoma, the relapse following treatment with MAPKi is driven by a small subpopulation of residual or “drug-tolerant” cells, termed minimal residual disease (MRD). MRD can contain four distinct drug-tolerant transcriptional states [13]. One of these states is characterized by high expression of neural crest stem cell (NCSC) markers. This NCSC subpopulation has a critical role in driving relapse.
The neural crest (NC) is a multipotent stem cell population generated during early vertebrate. NCSCs arise at the developing dorsal neural tube and have high migratory capacity [32]. These cells disseminate into the whole embryo to differentiate into a variety of cell types with different functions, such as peripheral nervous system cells, bone, cartilage, connective tissue, endocrine cells, and pigment cells. The acquisition of invasive phenotype and resistance is associated with gene regulatory networks reminiscent of this neural crest [32].
The retinoid X receptor gamma (RXRG) is the key driver of the NCSC state. Targeting the NCSC subpopulation using pan-RXR antagonist (HX531) enhances the antitumor activity of MAPKi, blocks the emergence of NCSCs, and prevents disease relapse of patient-derived melanoma [13]. Noteworthy, the pharmacological inhibition of RXRG lead to a concomitant increase in the invasive AXL-high subpopulation. Blocking the latter subpopulation is achievable and described in the section above.
All of the above findings illustrate the remarkable heterogeneity in melanoma and suggest that inhibition of these different invasive phenotypes is a very promising approach to overcome resistance and prevent relapse.
2.1.3. Targeting Ferroptosis to Block the Dedifferentiation Resistance Escape Route
Ferroptosis is a type of programmed cell death that occurs via an iron-dependent accumulation of lipids peroxides [33]. It is regulated by glutathione peroxidase 4 (GPX4). GPX4 is a selenoprotein that converts reduced glutathione (GSH) into oxidized glutathione (GSSG) and thereby reduces lipid peroxides (L-OOH). Inhibition of GPX4 activity leads to the accumulation of cytotoxic lipid peroxides, a hallmark of ferroptosis [33].
It was reported that the EMT-like or persister cell state in cancer cells is highly dependent on GPX4 activity for survival [34]. Accordingly, Tsoi et al. 2018 [14], in their study, showed that the undifferentiated phenotype frequently linked to innate and acquired resistance to targeted and immune therapies in melanoma is associated with increased sensitivity to ferroptosis inducing compounds, including erastin, ML162, and ML21s. Whereas the undifferentiated phenotype displays high sensitivity, the melanocytic state is the most resistant one.
Therefore, these findings support the rationale for combination strategies using ferroptosis-inducing drugs to block the dedifferentiation resistance escape route. Interestingly, there is new evidence demonstrating that ferroptosis-inducing therapy can enhance the effect of immune checkpoint inhibitors anti-PDL1 [33].
2.1.4. Targeting the Senescence-like Phenotype
The senescence-like phenotype is a form of tumor dormancy present in a subpopulation of tumor residual cells that escaped cell death following treatment [35]. Indeed, the senescence-like cells display resistance to apoptosis and secrete cytokines and growth factors referred as senescence-associated secretory phenotype (SASP) [35]. This SASP is associated with angiogenesis, invasion, and can promote metastasis [35]. The researchers previously showed that MAPKi can promote a senescence-like phenotype in melanoma cells [36]. Furthermore, the researchers identified that stressful microenvironmental conditions with high tyrosine induce a phenotypic switch towards an invasive or senescence-like state in melanoma primary cultures [37]. Altogether, these findings show that targeting the senescence-like phenotype is considered as an intriguing therapeutic strategy. Senolytics are a class of drugs that selectively eliminate senescent cells. These drugs include inhibitors of Bcl2 family, histone deacetylase inhibitors, inhibitors of PI3K/AKT/mTOR pathway, TKI, and flavonoids [16][17].
Bcl2 inhibitors, such as the BH3 mimetic navitoclax and the HDACi panobinostat, promote senescent cell apoptosis and are able to eliminate senescent cells that persist during chemotherapy [16][17]. Likewise, the combination of dasatinib and quercetin (Q + C) is also very promising in the clearance of senescent cells [16][17]. Navitoclax and the combination Q + C can also inhibit the SASP and reduce inflammation [16][17]. Moreover, the PI3K/AKT pathway is a main survival pathway in senescent cells. Thus, blocking the latter can be very promising, since PI3K/AKT inhibitors are investigated in clinic in several cancers [16][17].
Collectively, developing effective approaches to eliminate residual senescent-like cells in melanoma can be very promising to prevent relapse and inhibit metastasis. Nevertheless, exploring the best strategy and the optimal schedule of treatment is needed in future clinical applications.
2.1.5. Targeting Endothelin Receptor Signaling as a Unique Approach to Overcome Phenotypic Heterogeneity
The phenotypic heterogeneity is a key driver for therapy failure leading to the establishment of tumor states associated with acquired resistance and high metastatic potential.
Endothelin 1 (EDN1), which is implicated in several aspects of tumor progression [38], has been identified as a master regulator of phenotypic heterogeneity and can contribute to paracrine protection against MAPKi [15]. Indeed, EDN1 expression is upregulated under treatment and confers resistance through ERK reactivation. More important, EDN1 expression regulated by MITF can protect the proliferative phenotype characterized by MITFhigh via the endothelin receptor B (EDNRB), and it is crucial for the maintenance of the AXLhigh invasive phenotype via EDNRA [15].
The combination of EDNR antagonists, such as bosentan, macitentan, and BQ788, with MAPKi, lead to significant tumor growth and improve their response [15]. Targeting EDNR signaling not only inhibits distinct phenotypes but can also suppress a favorable microenvironment for tumor growth and progression through acting on cancer associated fibroblast and endothelial cells [38][15].
Therefore, targeting endothelin receptors can be a novel approach in melanoma for tackling phenotypic heterogeneity.
2.2. Targeting Lipid Metabolism as a New Strategy to Overcome Phenotypic Plasticity
Lipid metabolism contributes to melanoma plasticity and aggressiveness. Indeed, it regulates the dedifferentiation process and sustains cell growth and metastasis in melanoma [39][18]. Thus, targeting the lipid metabolic network emerges as an exciting new approach in cancer research and drug discovery.
Fatty acid (FA) synthesis is highly active in cancer cells and serves as a fuel source of energy that promotes tumor growth. Most of the enzymes associated with fatty acid synthesis (e.g., FASN, SCD, ACC, ACLY), and receptors related to lipid uptake (e.g., FAT/CD36, FATPs, and FABPpm) are upregulated in several cancers, including melanoma [39][18].
2.2.1. Blocking Fatty Acid Translocase (FAT/CD36)
CD36 is a transmembrane glycoprotein (also known as fatty acid translocase (FAT)) that has an essential role in FA uptake and is involved in metastasis and cancer cell growth [39][18]. In melanoma, the starved melanoma cell (SMC) state found early in minimal residual disease (MRD) is characterized by high expression of CD36 [13]. Accordingly, targeting metastasis-initiating cells by an anti-CD36 antibody showed high efficacy in oral cancer mouse models of human oral carcinomas [18].
Together, these findings show that blocking CD36 might be a promising strategy to prevent metastases and relapse in melanoma.
2.2.2. SCD Modulation
Stearoyl CoA desaturase 1 (SCD1) is an endoplasmic reticulum-associated lipogenic enzyme that converts saturated fatty acids (SFAs) into monounsaturated fatty acids (MUFA) [39][40]. The proportion of SFAs and MUFAs is involved in controlling cell proliferation and differentiation. Likewise, several studies reported an upregulation of SCD in several cancers and its implication in cell proliferation, migration, and metastasis during both early states and progression [39][40]. Therefore, SCD is considered a novel potential target in cancer therapy.
In melanoma, a strong correlation was found between SCD1 expression and the proliferative phenotype [39]. Vivas-García et al. 2020 showed that MITFhigh phenotype displays high sensitivity to SCD inhibitor consistent with a strong decrease in expression of genes implicated in cell cycle progression (E2F and MYC targets), whereas invasive/undifferentiated MITFlow phenotypes are insensitive to SCDi [39]. This undifferentiated state and thereby the metastatic capacity can be induced under SCDi as well as CD36 expression. Thus, MITFlow state may be more dependent on CD36 activity for the uptake of FAs from the microenvironment.
Altogether, these observations suggest that a therapeutic strategy that combines SCD inhibition and CD36 blockade would be very effective to refrain phenotypic plasticity in melanoma.
2.2.3. SREBPs Inhibition
Sterol regulatory element-binding protein-1 (SREBP-1) is a master regulator transcription factor of lipogenesis that regulates the expression of genes involved in lipid synthesis and uptake [41]. SREBP1 has an important role in cancer progression and metastasis. Several small-molecule inhibitors of SREBPs are available, such as fatostatin, botulin, and PF-429242. These inhibitors have demonstrated anti-tumor effects in various cancers in preclinical studies [19]. Importantly, SREBP-1 inhibition can sensitize resistant mutant BRAF melanoma both in vitro and in vivo models [42].
These findings suggest that targeting SREBP-1 can offer a new alternative to overcome acquired resistance in melanoma.
2.2.4. Fatty Acid Synthase (FASN) Inhibition
FASN is a central modulator of lipid metabolism catalyzing the last step in de novo-lipogenesis. It also plays a critical role in tumor growth and emerges as a target in cancer [20][21].
The early-generation FASN inhibitors, such as cerulenin and C75, have shown promising anticancer activity, but their pharmacological and off-target effects have limited their clinical development [20][21]. The recently developed TVB-2640 is a more specific inhibitor that is currently investigated in clinical trials in patients with solid tumors in combination with targeted therapy or chemotherapy. Cells with high lipogenic activity seem more sensitive to FASNi TVB-2640, so it can serve as selection criteria for patients that may likely benefit from FASNi therapy [20][21].
2.3. Targeting Glutamine Metabolism to Reduce Melanoma Plasticity and Aggressiveness
As glutamine metabolism develops in melanoma cells with acquired resistance to targeted therapies [43], it is believed that melanoma patients could benefit from treatment inhibiting glutamine entry into the cells or its consumption by the cells. Therefore, treatment combinations should be the best way to further induce cancer cell apoptosis.
2.3.1. Blocking Glutamine Import
Glutamine predominantly enters into the cells through the SLC1A5 transporter. Gamma-1-glutamyl-p-nitroanilide (GPNA) is a pharmacological inhibitor of SLC1A1, which has been synthetized [44] to block glutamine uptake. This inhibitor prevents tumor growth and induces apoptosis in lung cancer cells [45], but the GPNA effect has not been evaluated in melanoma yet. Another inhibitor of SLC1A1 is the benzyl serine, which has antiproliferative properties in melanoma cell lines [46].
2.3.2. Blocking Glutamine Use
BPTES (bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl) ethyl sulfide) is a molecule inhibiting the two GLS1 isoforms (GAC and KGA) but not the GLS2. BPTES induces a conformational change to form inactive tetramers. It has been used in vivo and in vitro in a model of BRAF mutated melanoma and reduces tumor growth [22]. However, this inhibitor cannot be used in clinic because of its low solubility in water. CB-839 is a new allosteric inhibitor of GLS1. This molecule has already shows antitumoral activities, both in vitro and in vivo in many cancer types, including melanoma [23]. This inhibitor can be used in clinic and is orally bioavailable. It is currently used in 22 phase I and phase II clinical trials, including one in melanoma (NCT02771626).
2.4. Targeting TME Components and Signals
2.4.1. Targeting Cancer-Associated Fibroblasts (CAFs) to Disrupt Melanoma Plasticity
Cancer-associated fibroblasts (CAFs) are critical components of the TME and are implicated in the regulation of cancer progression, immune crosstalk, and phenotypic heterogeneity and plasticity. Targeting cancer cell–CAF signaling interactions and proteins expressed by CAF are considered very attractive strategies [47][48].
CAF-secreted factors that promote phenotype switching in melanoma include TGF-β and FGFs. New anti-TGF-β agents have been developed and tested in clinical trials, including monoclonal antibodies, such as fresolimumab in combination with other therapies. These agents show encouraging efficacy and manageable tolerability [26].
The FGF/FGFR axis is also a promising therapeutic target in melanoma [49]. Several small-molecule inhibitors of FGFR receptors and other RTK (such as dovitinib) are available in clinics [27].
Moreover, fibroblast activation protein (FAP) is a proline selective serine protease that is overexpressed in various cancers and associated with worse prognosis [24]. FAP-targeted theranostics are small-molecule FAP inhibitors coupled with radiotracers for both diagnostic and therapeutic settings. Thus, FAPIs are very promising tracers for theranostic applications [24]. These molecules showed anti-cancer activity in preclinical studies [24][25] and they are investigated in several clinical trials [50].
Altogether, these findings show that CAF-targeted agents in combination with existing therapies could yield greater benefit in melanoma.
2.4.2. Targeting CD73 as a New Potential Therapeutic Opportunity
CD73 emerged as a regulator of melanoma plasticity [51] and can be considered an ideal target of cancer therapy. Indeed, CD73 is an immune inhibitory molecule that is upregulated in cancer and promotes metastases [52].
The safety and efficacy of multiple anti-CD73 monoclonal antibodies, such as oleclumab, CPI-006, and BMS-986179, are under investigation in clinical trials in combination with other therapies in multiple cancers [28]. Anti-CD73 also showed very promising results in melanoma, both in preclinical and clinical studies [29][30].
3. Conclusions
Despite recent advances in cancer treatment with the development of targeted therapies and immune checkpoint inhibitors, intratumoral heterogeneity and acquired resistance remain the major limitations. The development of resistance can be driven by both genetic and non-genetic mechanisms. Genetic mechanisms involve the emergence of preexisting clones containing specific genetic mutations, or the acquisition of de novo mutations. On the other hand, non-genetic mechanisms are mainly driven by phenotypic plasticity. In that regard, the natural selection theory (classical Darwinian selection) associated with somatic mutations and the Lamarckian induction concept associated with epigenetic and transcriptional reprogramming, contribute concurrently to phenotypic diversification and, thereby, drug resistance and metastases. In addition, the dynamic and the complex bidirectional interplay between cancer cells and the tumor microenvironment (TME) can govern tumor evolution. ROS can act as a critical mediator between TME signals and effectors of pathways regulating cancer phenotype. In melanoma, the significant advances in single cell RNA techniques and deep learning technologies allow for a detailed characterization and tracing of dynamic phenotypic changes and the identification of different trajectories followed by melanoma cells. Here, the research deepens the understanding of phenotype switching in melanoma and its master regulators. These regulators are involved in cell fate determination, not only in melanoma, but also in several cancer types. This switch towards adaptive phenotypes is orchestrated by the cooperativity of transcriptional, epigenetic, and metabolic networks. The increased knowledge of melanoma plasticity can provide crucial insight into many cancer types and better guide designs for novel therapeutic strategies. Here, the researchers proposed several rational treatment combinations having high potential clinical relevance that aim to target phenotype-specific vulnerabilities, including tumor microenvironments, with the aim to prevent further metastatic dissemination, overcome resistance and, therefore, improve patient outcome.
References
- Najem, A.; Krayem, M.; Perdrix, A.; Kerger, J.; Awada, A.; Journe, F.; Ghanem, G. New Drug Combination Strategies in Melanoma: Current Status and Future Directions. Anticancer Res. 2017, 37, 5941–5953.
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired Resistance and Clonal Evolution in Melanoma during BRAF Inhibitor Therapy. Cancer Discov. 2014, 4, 80–93.
- Grzywa, T.M.; Paskal, W.; Włodarski, P.K. Intratumor and Intertumor Heterogeneity in Melanoma. Transl. Oncol. 2017, 10, 956–975.
- Wouters, J.; Kalender-Atak, Z.; Minnoye, L.; Spanier, K.I.; De Waegeneer, M.; Bravo González-Blas, C.; Mauduit, D.; Davie, K.; Hulselmans, G.; Najem, A.; et al. Robust Gene Expression Programs Underlie Recurrent Cell States and Phenotype Switching in Melanoma. Nat. Cell Biol. 2020, 22, 986–998.
- Verfaillie, A.; Imrichova, H.; Atak, Z.K.; Dewaele, M.; Rambow, F.; Hulselmans, G.; Christiaens, V.; Svetlichnyy, D.; Luciani, F.; Van den Mooter, L.; et al. Decoding the Regulatory Landscape of Melanoma Reveals TEADS as Regulators of the Invasive Cell State. Nat. Commun. 2015, 6, 6683.
- Rambow, F.; Marine, J.-C.; Goding, C.R. Melanoma Plasticity and Phenotypic Diversity: Therapeutic Barriers and Opportunities. Genes Dev. 2019, 33, 1295–1318.
- Hoek, K.S.; Schlegel, N.C.; Brafford, P.; Sucker, A.; Ugurel, S.; Kumar, R.; Weber, B.L.; Nathanson, K.L.; Phillips, D.J.; Herlyn, M.; et al. Metastatic Potential of Melanomas Defined by Specific Gene Expression Profiles with No BRAF Signature. Pigment Cell Res. 2006, 19, 290–302.
- Boshuizen, J.; Koopman, L.A.; Krijgsman, O.; Shahrabi, A.; van den Heuvel, E.G.-; Ligtenberg, M.A.; Vredevoogd, D.W.; Kemper, K.; Kuilman, T.; Song, J.-Y.; et al. Cooperative Targeting of Melanoma Heterogeneity with an AXL Antibody-Drug Conjugate and BRAF/MEK Inhibitors. Nat. Med. 2018, 24, 203–212.
- Zhu, C.; Wei, Y.; Wei, X. AXL Receptor Tyrosine Kinase as a Promising Anti-Cancer Approach: Functions, Molecular Mechanisms and Clinical Applications. Mol. Cancer 2019, 18, 153.
- Loges, S.; Heuser, M.; Chromik, J.; Vigil C., E.; Paschka, P.; Re, F.; Renzo N., D.; Lemoli R., M.; Mattei, D.G.; Ben-Batalla, I.; et al. The Combination of AXL Inhibitor Bemcentinib and Low Dose Cytarabine Is Well Tolerated and Efficacious in Elderly Relapsed AML Patients: Update from the Ongoing BGBC003 Phase II Trial (NCT02488408). Blood 2020, 136, 14.
- Straume, O.; Lorens, J.B.; Gausdal, G.; Gjertsen, B.T.; Schuster, C. 1336TiP—A Randomized Phase Ib/II Study of the Selective Small Molecule Axl Inhibitor Bemcentinib (BGB324) in Combination with Either Dabrafenib/Trametinib (D/T) or Pembrolizumab in Patients with Metastatic Melanoma. Ann. Oncol. 2019, 30, v563.
- Sabbah, M.; Najem, A.; Krayem, M.; Awada, A.; Journe, F.; Ghanem, G.E. RTK Inhibitors in Melanoma: From Bench to Bedside. Cancers 2021, 13, 1685.
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855.e19.
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.L.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-Stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018, 33, 890–904.e5.
- Smith, M.P.; Rowling, E.J.; Miskolczi, Z.; Ferguson, J.; Spoerri, L.; Haass, N.K.; Sloss, O.; McEntegart, S.; Arozarena, I.; von Kriegsheim, A.; et al. Targeting Endothelin Receptor Signalling Overcomes Heterogeneity Driven Therapy Failure. EMBO Mol. Med. 2017, 9, 1011–1029.
- Kirkland, J.L.; Tchkonia, T. Senolytic Drugs: From Discovery to Translation. J. Intern. Med. 2020, 288, 518–536.
- Zhu, M.; Meng, P.; Ling, X.; Zhou, L. Advancements in Therapeutic Drugs Targeting of Senescence. Adv. Chronic Dis. 2020, 11, 2040622320964125.
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.-O.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting Metastasis-Initiating Cells through the Fatty Acid Receptor CD36. Nature 2017, 541, 41–45.
- Yang, J.; Stack, M.S. Lipid Regulatory Proteins as Potential Therapeutic Targets for Ovarian Cancer in Obese Women. Cancers 2020, 12, 3469.
- Fhu, C.W.; Ali, A. Fatty Acid Synthase: An Emerging Target in Cancer. Molecules 2020, 25, 3935.
- Koundouros, N.; Poulogiannis, G. Reprogramming of Fatty Acid Metabolism in Cancer. Br. J. Cancer 2020, 122, 4–22.
- Hernandez-Davies, J.E.; Tran, T.Q.; Reid, M.A.; Rosales, K.R.; Lowman, X.H.; Pan, M.; Moriceau, G.; Yang, Y.; Wu, J.; Lo, R.S.; et al. Vemurafenib Resistance Reprograms Melanoma Cells towards Glutamine Dependence. J. Transl. Med. 2015, 13, 210.
- Varghese, S.; Pramanik, S.; Williams, L.J.; Hodges, H.R.; Hudgens, C.W.; Fischer, G.M.; Luo, C.K.; Knighton, B.; Tan, L.; Lorenzi, P.L.; et al. The Glutaminase Inhibitor CB-839 (Telaglenastat) Enhances the Antimelanoma Activity of T-Cell-Mediated Immunotherapies. Mol. Cancer 2021, 20, 500–511.
- Lindner, T.; Loktev, A.; Giesel, F.; Kratochwil, C.; Altmann, A.; Haberkorn, U. Targeting of Activated Fibroblasts for Imaging and Therapy. EJNMMI Radiopharm. Chem. 2019, 4, 16.
- Watabe, T.; Liu, Y.; Kaneda-Nakashima, K.; Shirakami, Y.; Lindner, T.; Ooe, K.; Toyoshima, A.; Nagata, K.; Shimosegawa, E.; Haberkorn, U.; et al. Theranostics Targeting Fibroblast Activation Protein in the Tumor Stroma: 64Cu and 225Ac Labelled FAPI-04 in Pancreatic Cancer Xenograft Mouse Models. J. Nucl. Med. 2020, 61, 563–569.
- Ciardiello, D.; Elez, E.; Tabernero, J.; Seoane, J. Clinical Development of Therapies Targeting TGFβ: Current Knowledge and Future Perspectives. Ann. Oncol. 2020, 31, 1336–1349.
- Chandana, S.R.; Babiker, H.M.; Mahadevan, D. Clinical Complexity of Utilizing FGFR Inhibitors in Cancer Therapeutics. Expert Opin. Investig. Drugs 2020, 29, 1413–1429.
- Harvey, J.B.; Phan, L.H.; Villarreal, O.E.; Bowser, J.L. CD73’s Potential as an Immunotherapy Target in Gastrointestinal Cancers. Front. Immunol. 2020, 11, 508.
- Perrot, I.; Michaud, H.-A.; Giraudon-Paoli, M.; Augier, S.; Docquier, A.; Gros, L.; Courtois, R.; Déjou, C.; Jecko, D.; Becquart, O.; et al. Blocking Antibodies Targeting the CD39/CD73 Immunosuppressive Pathway Unleash Immune Responses in Combination Cancer Therapies. Cell Rep. 2019, 27, 2411–2425.e9.
- Soleimani, A.; Farshchi, H.K.; Mirzavi, F.; Zamani, P.; Ghaderi, A.; Amini, Y.; Khorrami, S.; Mashayekhi, K.; Jaafari, M.R. The Therapeutic Potential of Targeting CD73 and CD73-Derived Adenosine in Melanoma. Biochimie 2020, 176, 21–30.
- Boshuizen, J.; Pencheva, N.; Krijgsman, O.; Altimari, D.D.; Castro, P.G.; de Bruijn, B.; Ligtenberg, M.A.; den Heuvel, E.G.; Vredevoogd, D.W.; Song, J.-Y.; et al. Cooperative Targeting of Immunotherapy-Resistant Melanoma and Lung Cancer by an AXL-Targeting Antibody-Drug Conjugate and Immune Checkpoint Blockade. Cancer Res. 2021, 81, 1775–1787.
- Baggiolini, A.; Varum, S.; Mateos, J.M.; Bettosini, D.; John, N.; Bonalli, M.; Ziegler, U.; Dimou, L.; Clevers, H.; Furrer, R.; et al. Premigratory and Migratory Neural Crest Cells Are Multipotent in Vivo. Cell Stem Cell 2015, 16, 314–322.
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, Biology and Role in Disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282.
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-Tolerant Persister Cancer Cells Are Vulnerable to GPX4 Inhibition. Nature 2017, 551, 247–250.
- Saleh, T.; Tyutyunyk-Massey, L.; Gewirtz, D.A. Tumor Cell Escape from Therapy-Induced Senescence as a Model of Disease Recurrence after Dormancy. Cancer Res. 2019, 79, 1044–1046.
- Krayem, M.; Najem, A.; Journe, F.; Morandini, R.; Sales, F.; Awada, A.; Ghanem, G.E. Acquired Resistance to BRAFi Reverses Senescence-like Phenotype in Mutant BRAF Melanoma. Oncotarget 2018, 9, 31888–31903.
- Najem, A.; Wouters, J.; Krayem, M.; Rambow, F.; Sabbah, M.; Sales, F.; Awada, A.; Aerts, S.; Journe, F.; Marine, J.-C.; et al. Tyrosine-Dependent Phenotype Switching Occurs Early in Many Primary Melanoma Cultures Limiting Their Translational Value. Front. Oncol. 2021, 11, 780654.
- Bagnato, A.; Spinella, F.; Rosanò, L. The Endothelin Axis in Cancer: The Promise and the Challenges of Molecularly Targeted Therapy. Can. J. Physiol. Pharm. 2008, 86, 473–484.
- Vivas-García, Y.; Falletta, P.; Liebing, J.; Louphrasitthiphol, P.; Feng, Y.; Chauhan, J.; Scott, D.A.; Glodde, N.; Chocarro-Calvo, A.; Bonham, S.; et al. Lineage-Restricted Regulation of SCD and Fatty Acid Saturation by MITF Controls Melanoma Phenotypic Plasticity. Mol. Cell 2020, 77, 120–137.e9.
- Oatman, N.; Dasgupta, N.; Arora, P.; Choi, K.; Gawali, M.V.; Gupta, N.; Parameswaran, S.; Salomone, J.; Reisz, J.A.; Lawler, S.; et al. Mechanisms of Stearoyl CoA Desaturase Inhibitor Sensitivity and Acquired Resistance in Cancer. Sci. Adv. 2021, 7, eabd7459.
- Pellerin, L.; Carrié, L.; Dufau, C.; Nieto, L.; Ségui, B.; Levade, T.; Riond, J.; Andrieu-Abadie, N. Lipid Metabolic Reprogramming: Role in Melanoma Progression and Therapeutic Perspectives. Cancers 2020, 12, 3147.
- Talebi, A.; Dehairs, J.; Rambow, F.; Rogiers, A.; Nittner, D.; Derua, R.; Vanderhoydonc, F.; Duarte, J.A.G.; Bosisio, F.; Van den Eynde, K.; et al. Sustained SREBP-1-Dependent Lipogenesis as a Key Mediator of Resistance to BRAF-Targeted Therapy. Nat. Commun. 2018, 9, 2500.
- Soumoy, L.; Schepkens, C.; Krayem, M.; Najem, A.; Tagliatti, V.; Ghanem, G.E.; Saussez, S.; Colet, J.-M.; Journe, F. Metabolic Reprogramming in Metastatic Melanoma with Acquired Resistance to Targeted Therapies: Integrative Metabolomic and Proteomic Analysis. Cancers 2020, 12, 1323.
- Esslinger, C.S.; Cybulski, K.A.; Rhoderick, J.F. Ngamma-Aryl Glutamine Analogues as Probes of the ASCT2 Neutral Amino Acid Transporter Binding Site. Bioorg. Med. Chem. 2005, 13, 1111–1118.
- Hassanein, M.; Qian, J.; Hoeksema, M.D.; Wang, J.; Jacobovitz, M.; Ji, X.; Harris, F.T.; Harris, B.K.; Boyd, K.L.; Chen, H.; et al. Targeting SLC1a5-Mediated Glutamine Dependence in Non-Small Cell Lung Cancer. Int. J. Cancer 2015, 137, 1587–1597.
- Wang, Q.; Beaumont, K.A.; Otte, N.J.; Font, J.; Bailey, C.G.; van Geldermalsen, M.; Sharp, D.M.; Tiffen, J.C.; Ryan, R.M.; Jormakka, M.; et al. Targeting Glutamine Transport to Suppress Melanoma Cell Growth. Int. J. Cancer 2014, 135, 1060–1071.
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A Framework for Advancing Our Understanding of Cancer-Associated Fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186.
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.C. Targeting the Tumour Stroma to Improve Cancer Therapy. Nat. Rev. Clin. Oncol 2018, 15, 366–381.
- Grimm, J.; Hufnagel, A.; Wobser, M.; Borst, A.; Haferkamp, S.; Houben, R.; Meierjohann, S. BRAF Inhibition Causes Resilience of Melanoma Cell Lines by Inducing the Secretion of FGF1. Oncogenesis 2018, 7, 71.
- Windisch, P.; Zwahlen, D.R.; Koerber, S.A.; Giesel, F.L.; Debus, J.; Haberkorn, U.; Adeberg, S. Clinical Results of Fibroblast Activation Protein (FAP) Specific PET and Implications for Radiotherapy Planning: Systematic Review. Cancers 2020, 12, 2629.
- Reinhardt, J.; Landsberg, J.; Schmid-Burgk, J.L.; Ramis, B.B.; Bald, T.; Glodde, N.; Lopez-Ramos, D.; Young, A.; Ngiow, S.F.; Nettersheim, D.; et al. MAPK Signaling and Inflammation Link Melanoma Phenotype Switching to Induction of CD73 during Immunotherapy. Cancer Res. 2017, 77, 4697–4709.
- Antonioli, L.; Yegutkin, G.G.; Pacher, P.; Blandizzi, C.; Haskó, G. Anti-CD73 in Cancer Immunotherapy: Awakening New Opportunities. Trends Cancer 2016, 2, 95–109.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
743
Revisions:
6 times
(View History)
Update Date:
13 Apr 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No