 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Arianna Casciati | + 4823 word(s) | 4823 | 2022-03-17 04:42:32 | | | |
| 2 | Rita Xu | -1626 word(s) | 3197 | 2022-03-29 11:02:31 | | | | |
| 3 | Rita Xu | Meta information modification | 3197 | 2022-03-31 04:48:35 | | |
Video Upload Options
Glioblastoma multiforme (GBM) is the most common brain cancer in adults. GBM starts from a small fraction of poorly differentiated and aggressive cancer stem cells (CSCs) responsible for aberrant proliferation and invasion. Due to extreme tumor heterogeneity, actual therapies provide poor positive outcomes, and cancers usually recur. Therefore, alternative approaches, possibly targeting CSCs, are necessary against GBM.
1. Introduction
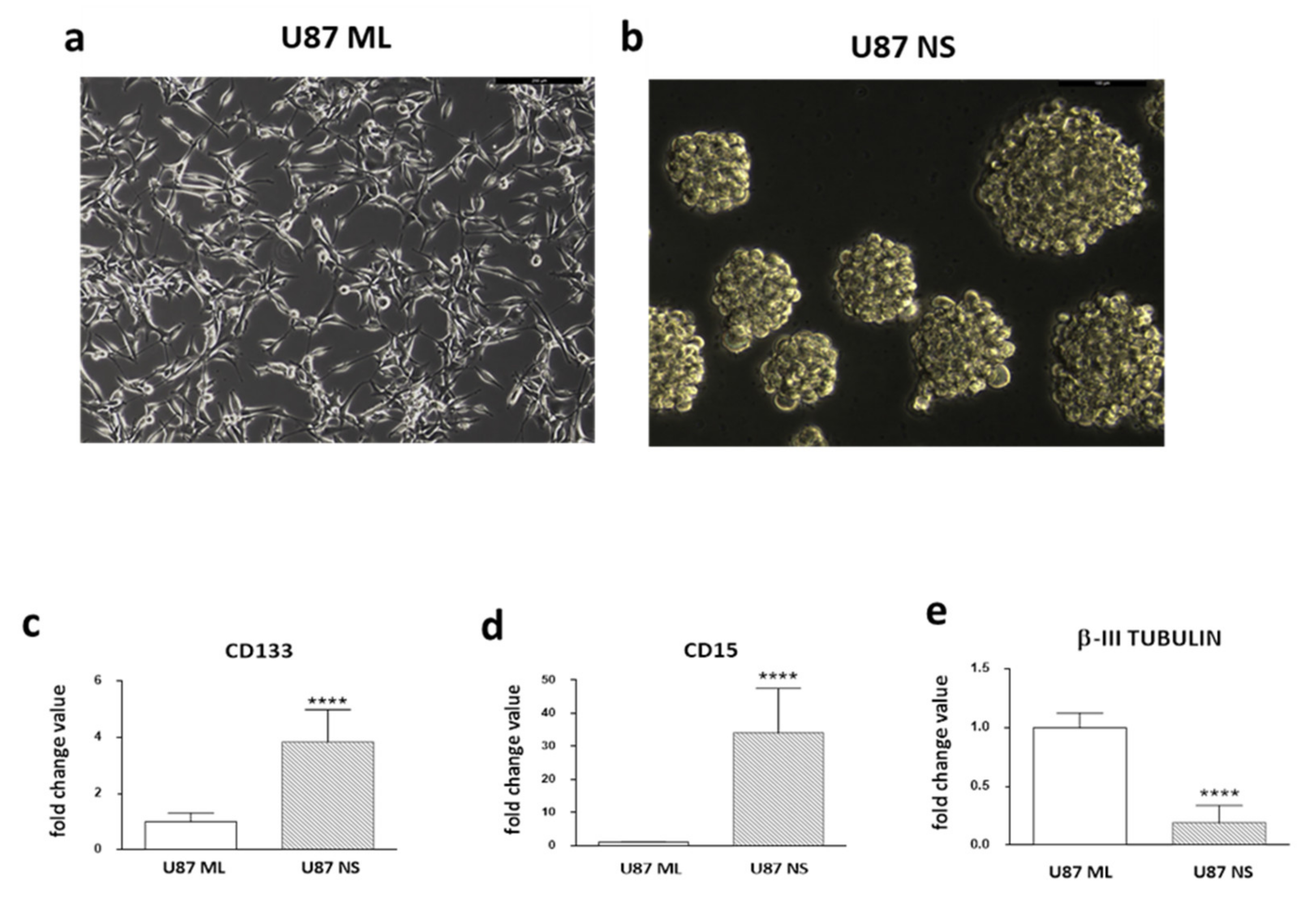
2. Expression Pattern of Stemness/Differentiation Genes in U87 ML and U87 NS Cells
3. Membrane Permeabilization and ROS Production after PEF-5 Exposure
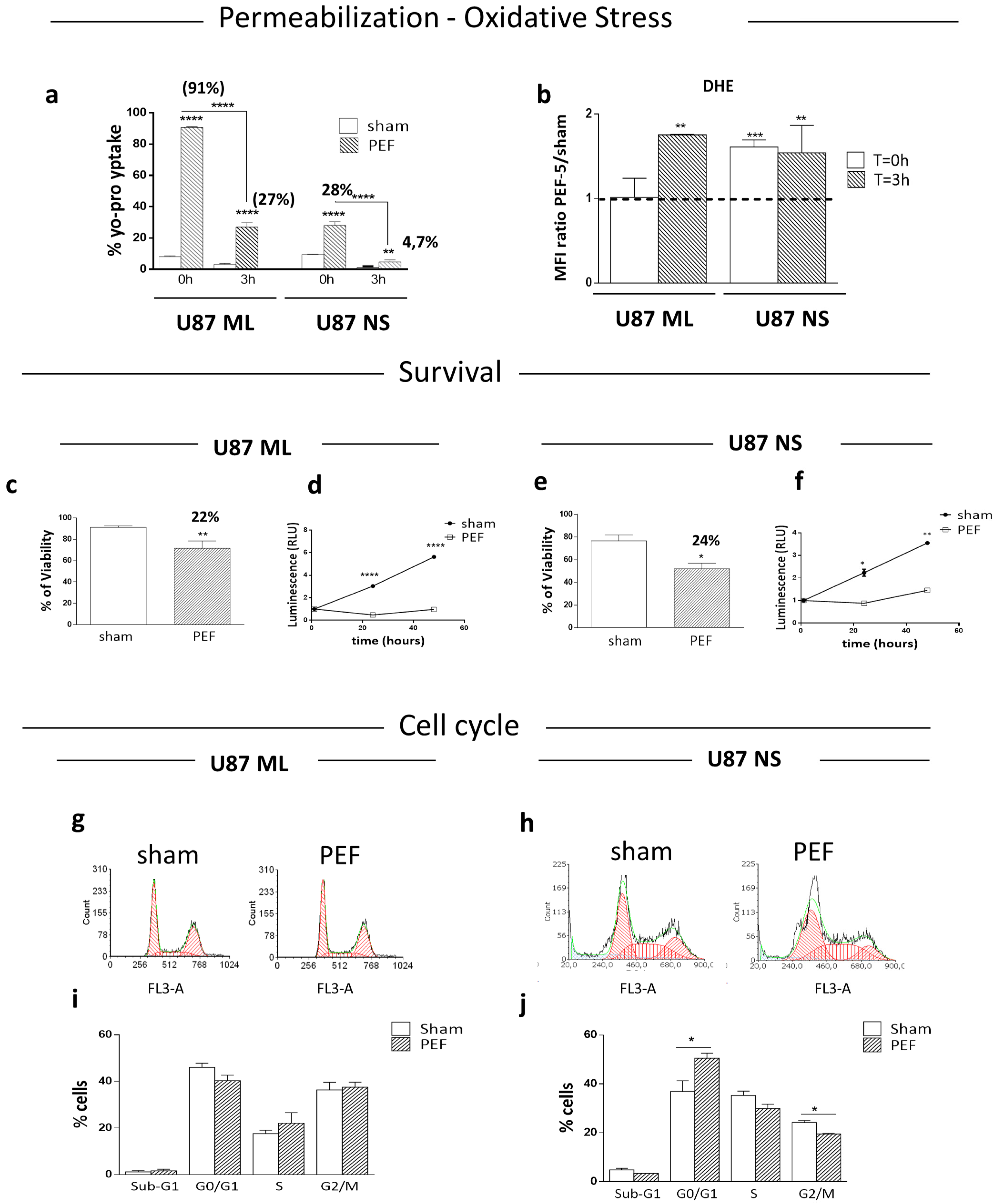
4. Survival, Proliferation and Cell Cycle Perturbation
5. Transcriptomic Profiles of U87 ML and NS Cells and Validation of mRNA Expression
5.1. Affymetrix Gene Chip Analyses
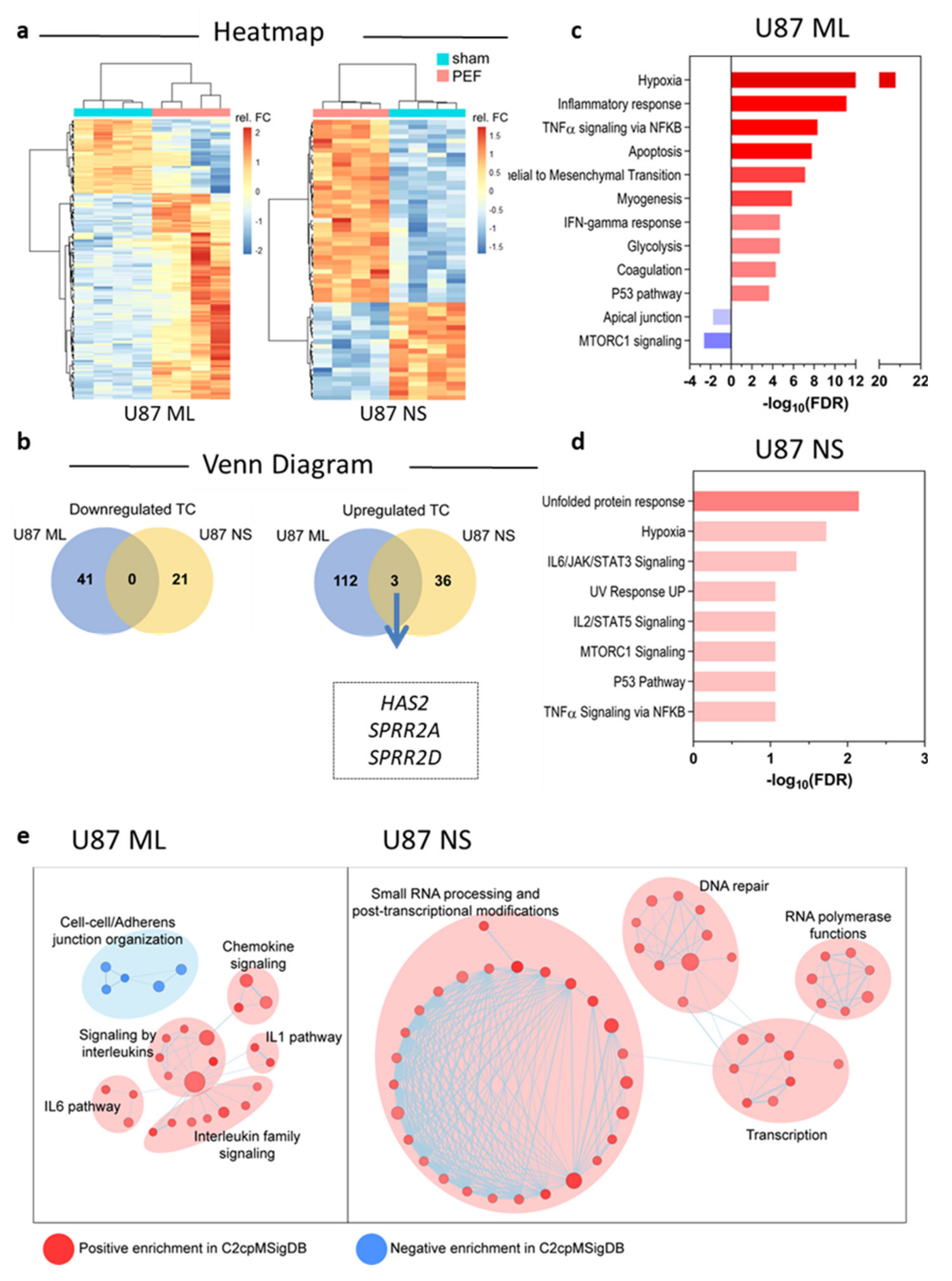
5.2. qPCR Validation of Transcriptome Data for Selected Genes
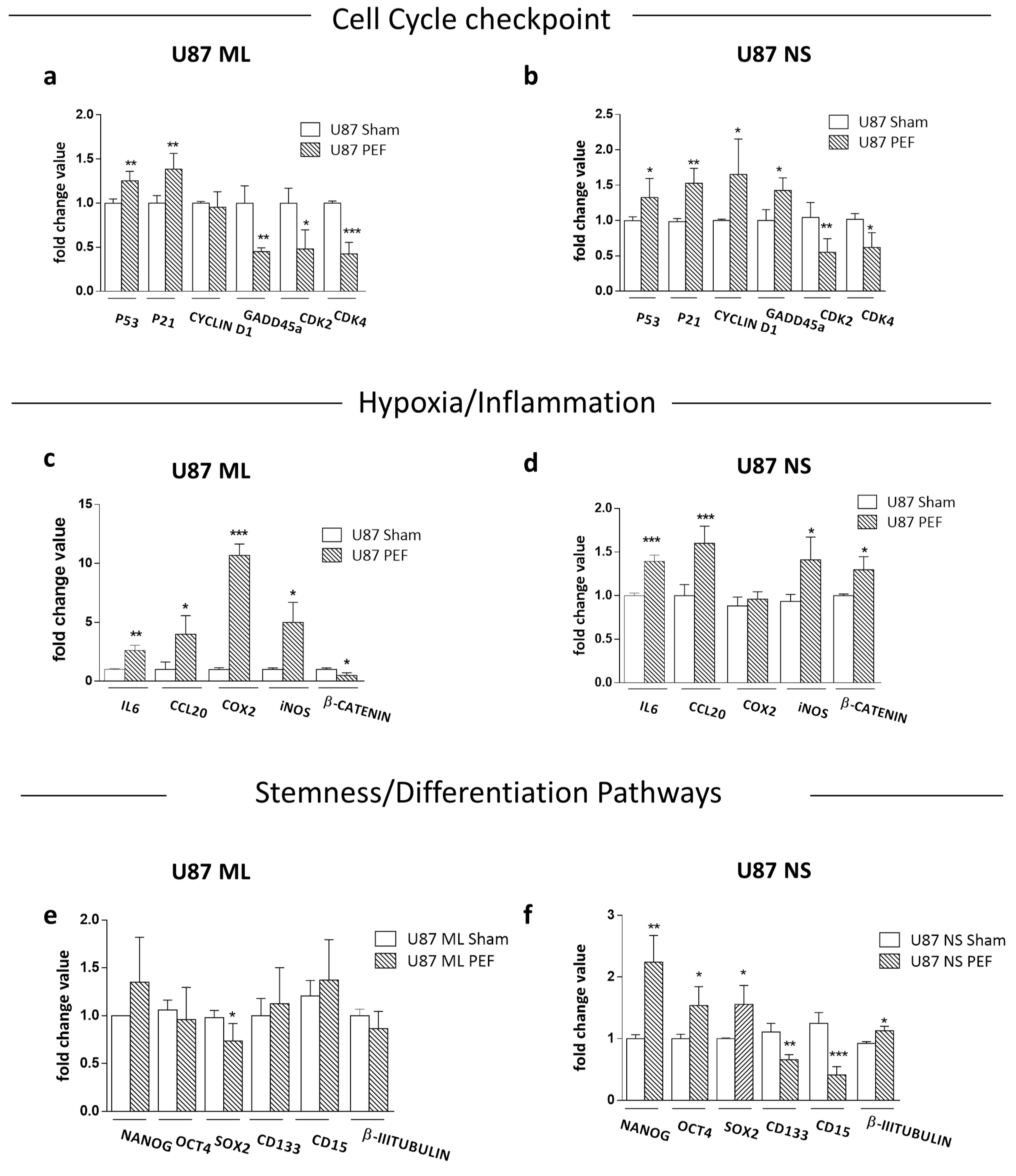
5.3. Effects of PEF Exposure on the Stemness/Differentiation Status of GBM Cells
6. Functional testing after PEF-5 exposure
6.1. Clonogenic survival assay and IR combined treatment
To assess possible influence of PEF-5 on GBM clonogenic capacity, the survival clonogenic assay was performed on both first neurospheres derived from U87 ML (U87 NS1) and second generation neurospheres, derived from U87 NS cells (U87 NS2). As shown in Figure 5, exposure to PEF-5 efficiently decreased the clonogenic capacity in both neurosphere generations (about 2-fold lower than sham-exposed cells; p < 0.0001 in U87 ML and p < 0.001 in U87 NS). These results again suggested a strong ability of PEF-5 to modify the stem phenotype of CSCs decreasing their self-renewal capacity (Figure 5a-d). As radiosensitivity increases with cell differentiation status, researchers tested the effectiveness of a combined protocol, irradiating U87 ML and U87 NS cells with increasing IR doses (2, 5, 8 Gy), 3 hours after PEF-5 exposure, as suggested in the previous work. As shown in Figure 5a-d, the combined treatment of PEF-5 and IR was able to reduce clone formation as function of delivered doses, suggesting an additive effect between these two physical agents.
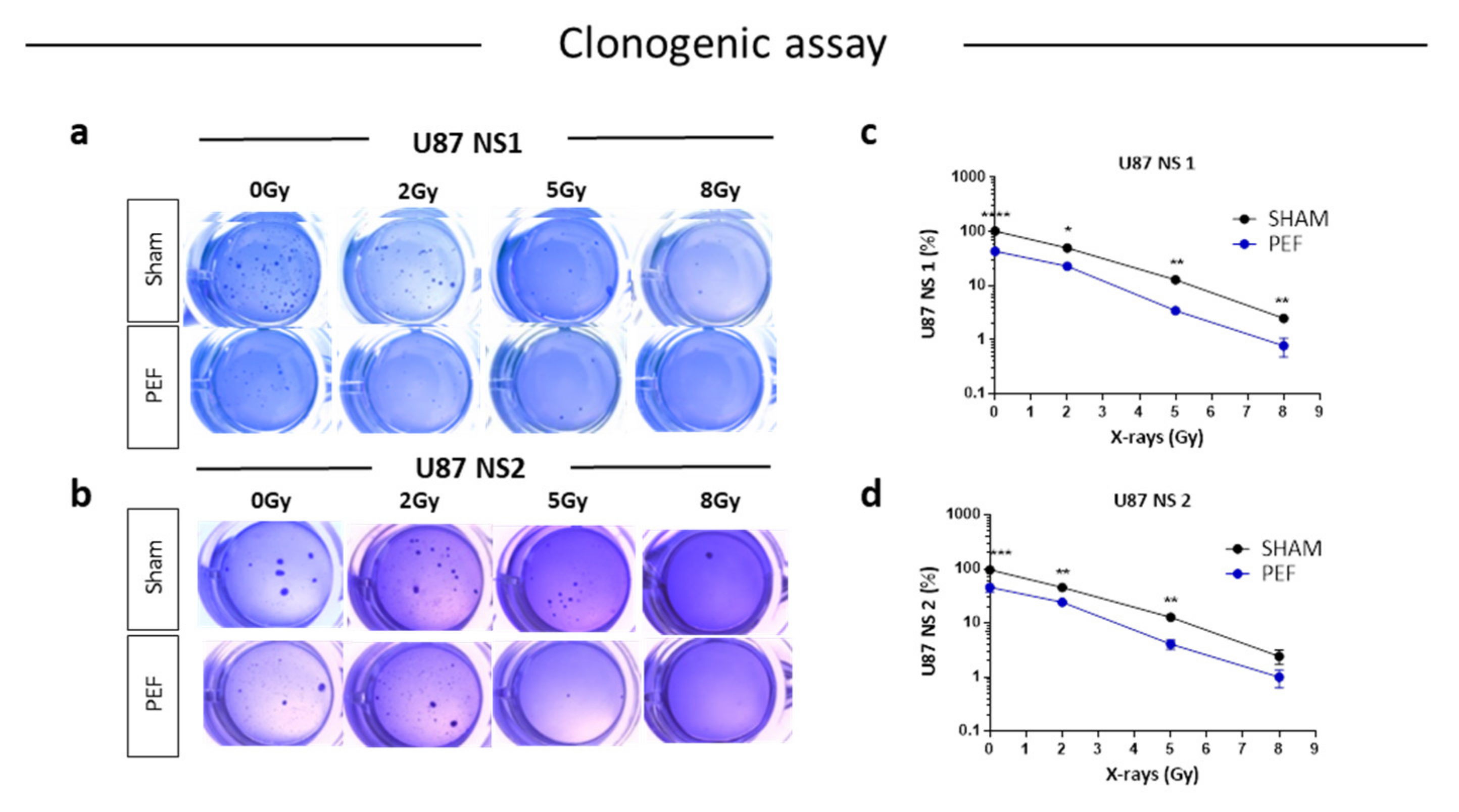
Figure 5. Survival clonogenic assay. (a-b) Representative images of primary and secondary neurospheres after PEF-5 exposure and IR treatments. (c-d) Quantitative analysis revealed a significant decrease in the clonogenic capacity between sham- and PEF-5 exposed cells, regardless of culture conditions. The combined treatment (PEF-5 + IR) reduced clone formation as function of delivered radiation doses.
6.2. Invasion assay after PEF-5 exposure
As invasiveness is another pathophysiological feature of human malignant gliomas, the effects of PEF-5 on the invasiveness and migration of GBM cells were checked in vitro by Transwell Matrigel invasion assay. As shown in Figure 6(a-d), the invasiveness of U87 ML and NS cells exposed to PEF-5 was strongly reduced compared with the control groups. U87 ML PEF-5 exposed cells showed a corresponding decrease of the invasion of 34% and U87 NS cells of 47%. This finding indicates the capacity of PEF-5 alone to inhibit the invasive ability of GBM cells in vitro. Also, for this assay, a combined protocol, PEF-5 + IR, was carried out using increasing IR doses (2, 5, 8 Gy) delivered 3 h after PEF-5 treatment. No further decrease in invasion ability was observed in only irradiated U87 ML samples (Figure 6c), instead, a decrease of 28% (p = 0.006) and of 29% (p = 0.009) was observed in U87 NS cells irradiated with 5 and 8 Gy, respectively (Figure 6d). This result may point out a migration response mediated by the cell types, and the specific delivered radiation dose. After the combined protocol of exposure, no significant changes were observed in migration and invasion abilities of these two cell types compared to those observed in the PEF-5 only treated groups. This result highlights the radio-resistance of GBM cells in both culture conditions. Notably, PEF-5 treatment alone was as effective as the combined treatment with radiations at all the delivered doses in both U87 ML (PEF vs 2Gy, -41% p = 0.04; PEF vs 5Gy, -32% p=0.0096; PEF vs 8Gy, -25% p=0.016), and U87 NS cells (PEF vs 2Gy, -41% p = 0.04).
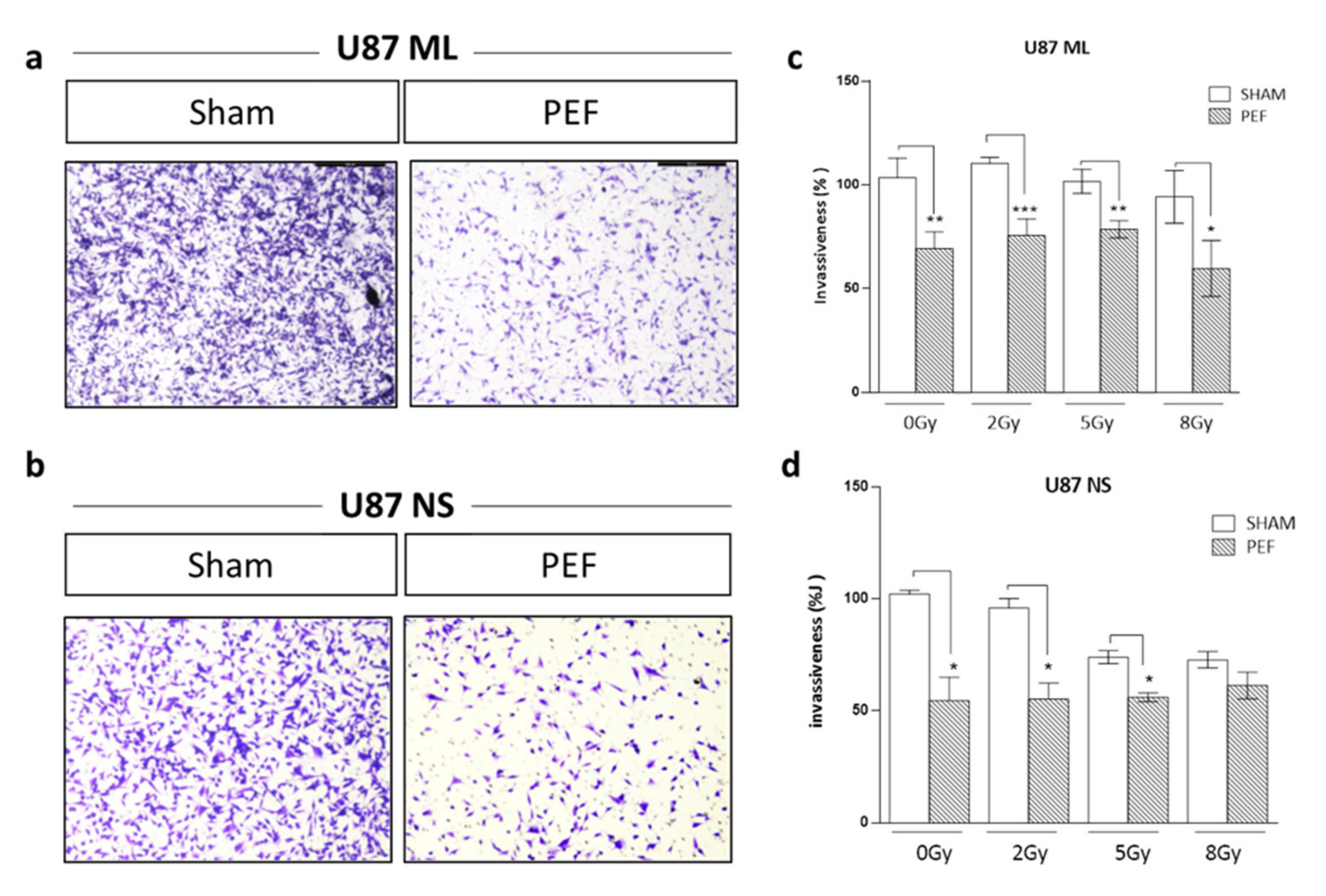
Figure 6. Invasion and migration assay. Representative images of transmigrated cells for (a) U87 ML and (b) U87 NS cells. (c-d) Percentage of invasiveness in U87 ML and NS cells evaluated 24 hours after PEF-5 exposure and IR combined treatment.
References
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.; Lightner, D.D.; Barnholtz-Sloan, J.; Villano, J.L. Epidemiologic and Molecular Prognostic Review of Glioblastoma. Cancer Epidemiol. Biomarkers Prev. 2014, 23, 1985–1996.
- Bonavia, R.; Inda, M.-d.-M.; Cavenee, W.K.; Furnari, F.B. Heterogeneity Maintenance in Glioblastoma: A Social Network. Cancer Res. 2011, 71, 4055–4060.
- Marusyk, A.; Polyak, K. Tumor heterogeneity: Causes and consequences. Biochim. Biophys. Acta 2010, 1805, 105–117.
- Meyer, M.; Reimand, J.; Lan, X.; Head, R.; Zhu, X.; Kushida, M.; Bayani, J.; Pressey, J.C.; Lionel, A.C.; Clarke, I.D.; et al. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc. Natl. Acad. Sci. USA 2015, 112, 851–856.
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401.
- Snuderl, M.; Fazlollahi, L.; Le, L.P.; Nitta, M.; Zhelyazkova, B.H.; Davidson, C.J.; Akhavanfard, S.; Cahill, D.; Aldape, K.D.; Betensky, R.A.; et al. Mosaic Amplification of Multiple Receptor Tyrosine Kinase Genes in Glioblastoma. Cancer Cell 2011, 20, 810–817.
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014.
- Szerlip, N.J.; Pedraza, A.; Chakravarty, D.; Azim, M.; McGuire, J.; Fang, Y.; Ozawa, T.; Holland, E.C.; Huse, J.T.; Jhanwar, S.; et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc. Natl. Acad. Sci. USA 2012, 109, 3041–3046.
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; DiMeco, F.; Vescovi, A. Isolation and Characterization of Tumorigenic, Stem-like Neural Precursors from Human Glioblastoma. Cancer Res. 2004, 64, 7011–7021.
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828.
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401.
- Ivey, J.W.; Wasson, E.M.; Alinezhadbalalami, N.; Kanitkar, A.; Debinski, W.; Sheng, Z.; Davalos, R.V.; Verbridge, S.S. Characterization of Ablation Thresholds for 3D-Cultured Patient-Derived Glioma Stem Cells in Response to High-Frequency Irreversible Electroporation. Research 2019, 2019, 8081315.
- Jordan, C.T.; Guzman, M.L.; Noble, M. Cancer Stem Cells. N. Engl. J. Med. 2006, 355, 1253–1261.
- Auffinger, B.; Tobias, A.L.; Han, Y.; Lee, G.; Guo, D.; Dey, M.; Lesniak, M.S.; Ahmed, A.U. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014, 21, 1119–1131.
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20.
- Flemming, A. Targeting the root of cancer relapse. Nat. Rev. Drug Discov. 2015, 14, 165.
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A "state of the science" review. Neuro Oncol. 2014, 16, 896–913.
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111.
- Jolly, M.K.; Celià-Terrassa, T. Dynamics of Phenotypic Heterogeneity Associated with EMT and Stemness during Cancer Progression. J. Clin. Med. 2019, 8, 1542.
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955.
- Gupta, P.B.; Fillmore, C.M.; Jiang, G.; Shapira, S.D.; Tao, K.; Kuperwasser, C.; Lander, E.S. Stochastic State Transitions Give Rise to Phenotypic Equilibrium in Populations of Cancer Cells. Cell 2011, 146, 633–644.
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715.
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45.
- Cornelis, F.H.; Ben Ammar, M.; Nouri-Neuville, M.; Matton, L.; Benderra, M.A.; Gligorov, J.; Fallet, V.; Mir, L.M. Percutaneous Image-Guided Electrochemotherapy of Spine Metastases: Initial Experience. Cardiovasc. Interv. Radiol. 2019, 42, 1806–1809.
- Davalos, R.V.; Mir, L.M.; Rubinsky, B. Tissue Ablation with Irreversible Electroporation. Ann. Biomed. Eng. 2005, 33, 223–231.
- Gehl, J.; Sersa, G.; Matthiessen, L.W.; Muir, T.; Soden, D.; Occhini, A.; Quaglino, P.; Curatolo, P.; Campana, L.G.; Kunte, C.; et al. Updated standard operating procedures for electrochemotherapy of cutaneous tumours and skin metastases. Acta Oncol. 2018, 57, 874–882.
- Sano, M.; Arena, C.B.; Bittleman, K.R.; DeWitt, M.R.; Cho, H.J.; Szot, C.S.; Saur, D.; Cissell, J.M.; Robertson, J.; Lee, Y.W.; et al. Bursts of Bipolar Microsecond Pulses Inhibit Tumor Growth. Sci. Rep. 2015, 5, 14999.
- Al-Sakere, B.; André, F.; Bernat, C.; Connault, E.; Opolon, P.; Davalos, R.V.; Rubinsky, B.; Mir, L.M. Tumor Ablation with Irreversible Electroporation. PLoS ONE 2007, 2, e1135.
- Kotnik, T.; Rems, L.; Tarek, M.; Miklavčič, D. Membrane Electroporation and Electropermeabilization: Mechanisms and Models. Annu. Rev. Biophys. 2019, 48, 63–91.
- Yarmush, M.L.; Golberg, A.; Serša, G.; Kotnik, T.; Miklavčič, D. Electroporation-Based Technologies for Medicine: Principles, Applications, and Challenges. Annu. Rev. Biomed. Eng. 2014, 16, 295–320.
- Arena, C.B.; Garcia, P.A.; Sano, M.B.; Olson, J.D.; Rogers-Cotrone, T.; Rossmeisl, J.H.; Davalos, R.V. Focal blood-brain-barrier disruption with high-frequency pulsed electric fields. Technology 2014, 2, 206–213.
- Garcia, P.A.; Rossmeisl, J.H.; Robertson, J.L.; Olson, J.D.; Johnson, A.J.; Ellis, T.L.; Davalos, R.V. 7.0-T Magnetic Resonance Imaging Characterization of Acute Blood-Brain-Barrier Disruption Achieved with Intracranial Irreversible Electroporation. PLoS ONE 2012, 7, e50482.
- Ringel-Scaia, V.M.; Beitel-White, N.; Lorenzo, M.F.; Brock, R.; Huie, K.E.; Coutermarsh-Ott, S.; Eden, K.; McDaniel, D.; Verbridge, S.S.; Rossmeisl, J.H.; et al. High-frequency irreversible electroporation is an effective tumor ablation strategy that induces immunologic cell death and promotes systemic anti-tumor immunity. EBioMedicine 2019, 44, 112–125.
- Tanori, M.; Casciati, A.; Zambotti, A.; Pinto, R.; Gianlorenzi, I.; Pannicelli, A.; Giardullo, P.; Benassi, B.; Marino, C.; Mancuso, M.; et al. Microsecond Pulsed Electric Fields: An Effective Way to Selectively Target and Radiosensitize Medulloblastoma Cancer Stem Cells. Int. J. Radiat. Oncol. 2021, 109, 1495–1507.
- Mizuguchi, Y.; Specht, S.; Lunz, J.G.; Isse, K.; Corbitt, N.; Takizawa, T.; Demetris, A.J. SPRR2A enhances p53 deacetylation through HDAC1 and down regulates p21 promoter activity. BMC Mol. Biol. 2012, 13, 20.
- Parnigoni, A.; Caon, I.; Moretto, P.; Viola, M.; Karousou, E.; Passi, A.; Vigetti, D. The role of the multifaceted long non-coding RNAs: A nuclear-cytosolic interplay to regulate hyaluronan metabolism. Matrix Biol. Plus 2021, 11, 100060.
- Yoo, K.-C.; Suh, Y.; An, Y.; Lee, H.-J.; Jeong, Y.J.; Uddin, N.; Cui, Y.-H.; Roh, T.H.; Shim, J.-K.; Chang, J.H.; et al. Proinvasive extracellular matrix remodeling in tumor microenvironment in response to radiation. Oncogene 2018, 37, 3317–3328.
- Zou, M.; Zhu, W.; Wang, L.; Shi, L.; Gao, R.; Ou, Y.; Chen, X.; Wang, Z.; Jiang, A.; Liu, K.; et al. AEG-1/MTDH-activated autophagy enhances human malignant glioma susceptibility to TGF-β1-triggered epithelial-mesenchymal transition. Oncotarget 2016, 7, 13122–13138.
- Orjalo, A.V.; Bhaumik, D.; Gengler, B.K.; Scott, G.K.; Campisi, J. Cell surface-bound IL-1α is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc. Natl. Acad. Sci. USA 2009, 106, 17031–17036.
- Boso, D.; Rampazzo, E.; Zanon, C.; Bresolin, S.; Maule, F.; Porcù, E.; Cani, A.; Della Puppa, A.; Trentin, L.; Basso, G.; et al. HIF-1α/Wnt signaling-dependent control of gene transcription regulates neuronal differentiation of glioblastoma stem cells. Theranostics 2019, 9, 4860–4877.
- Fitzpatrick, S.F.; Tambuwala, M.M.; Bruning, U.; Schaible, B.; Scholz, C.C.; Byrne, A.; O’Connor, A.; Gallagher, W.M.; Lenihan, C.R.; Garvey, J.F.; et al. An Intact Canonical NF-κB Pathway Is Required for Inflammatory Gene Expression in Response to Hypoxia. J. Immunol. 2010, 186, 1091–1096.

