DNA replication stress is a constant threat that cells must manage to proliferate and maintain genome integrity. DNA replication stress responses, a subset of the broader DNA damage response (DDR), operate when the DNA replication machinery (replisome) is blocked or replication forks collapse during S phase. There are many sources of replication stress, such as DNA lesions caused by endogenous and exogenous agents including commonly used cancer therapeutics, and difficult-to-replicate DNA sequences comprising fragile sites, G-quadraplex DNA, hairpins at trinucleotide repeats, and telomeres. Replication stress is also a consequence of conflicts between opposing transcription and replication, and oncogenic stress which dysregulates replication origin firing and fork progression. Cells initially respond to replication stress by protecting blocked replisomes, but if the offending problem (e.g., DNA damage) is not bypassed or resolved in a timely manner, forks may be cleaved by nucleases, inducing a DNA double-strand break (DSB) and providing a means to accurately restart stalled forks via homologous recombination. However, DSBs pose their own risks to genome stability if left unrepaired or misrepaired.
1. Introduction
Accurate DNA replication and proper chromosome segregation to daughter cells are critical to maintaining genome integrity and preventing cancer. Replication of the 6.3 billion bp of the diploid human genome during a typical eight-hour S phase requires >30,000 active origins, ~5000 of which are active at a time
[1][2]. Replication forks travel in a highly processive manner, synthesizing ~3000 bp per min, yet forks frequently encounter obstacles that stall replisomes, causing replication stress and triggering stress responses including the intra-S checkpoint
[3], fork protection to prevent replisome dissociation or fork collapse, and repair mechanisms that restart damaged forks. Replication stress is caused by a wide variety of endogenous and exogenous factors. Spontaneous DNA damage is caused by endogenous reactive oxygen species formed during cellular metabolism
[4][5], misincorporation of ribonucleotides, and DNA lability
[5]. DNA damage is also caused by exogenous genotoxic chemicals, and by ionizing and non-ionizing radiation. The vast majority of DNA lesions block replicative polymerases, necessitating lesion repair by an appropriate repair pathway, lesion bypass (damage tolerance) by translesion DNA synthesis (TLS) polymerases, repriming, homologous recombination (HR) mediated template switching, or passive rescue from an adjacent fork
[6][7]. DNA lesions that block replicative polymerases include nucleotides with broken rings, oxidized bases, and chemical adducts, as well as single- and double-strand breaks (DSBs). DNA polymerase inhibitors and depletion of nucleotide pools with hydroxyurea are exogenous sources that cause global replication stress, slowing or stopping most or all replication forks
[8].
Additional endogenous sources of replication stress are difficult to replicate DNA sequences and certain chromatin environments (e.g., G-quadraplex DNA, common fragile sites, telomeric DNA)
[9][10][11][12][13][14][15][16][17]. Replication stress is also caused by stable R-loops which form by hybridization of RNA transcripts to DNA templates, especially in G-rich sequences
[18][19][20], and by collisions between opposing transcription and replication machinery, particularly in highly transcribed ribosomal RNA gene arrays, fragile sites, and telomeres
[21][22][23][24][25][26][27]. Topoisomerases avert replication stress by preventing DNA overwinding in front of replication forks, a type of intrinsic, topological replication stress. A recent yeast study showed that cohesin, a highly conserved protein with essential roles in sister chromatid cohesion required for proper chromosome segregation, increases replication stress in centromeric and ribosomal DNA by trapping topological stress
[28]. Although cells suffer replication stress at random sites throughout the genome due to spontaneous (or induced) DNA damage, the stress associated with difficult to replicate sequences and challenging chromatin environments must be managed at those sites in every S phase.
When replication forks are blocked, the initial response has two aims: (1) protect the replication fork by stabilizing the replisome machinery, and (2) protect the fork from nucleolytic attack
[29][30]. If a blocked fork is not restarted in a timely manner, it may be cleaved by structure-specific nucleases yielding a single-ended DSB (seDSB) that is processed by resection nucleases to suppress misrepair by canonical non-homologous end-joining (cNHEJ) and promote accurate fork restart by HR. This is important because cNHEJ is the dominant DSB repair pathway in mammalian cells
[31][32] and cNHEJ of seDSBs can cause deletions and translocations that produce acentric and dicentric chromosomes that segregate improperly in mitosis or induce breakage-bridge-fusion cycles that further threaten genome integrity
[33].
2. DDR Signaling in Response to Replication Stress
DNA repair and DNA damage checkpoint systems minimize replication fork encounters with blocking lesions
[34][35][36], but with a steady state of ~10,000 DNA lesions per cell in unstressed cells
[5] and ~5000 simultaneously active replisomes during S phase
[2], fork encounters with blocking lesions are unavoidable. Acute or chronic exposures to genotoxic chemicals and radiation greatly increase replication stress, as does dysregulated replication associated with oncogenic stress
[37][38][39]. Under normal circumstances, the leading and lagging strand replication machines are coupled, traveling together with the MCM (minichromosome maintenance) replicative helicase. If the leading strand polymerase is blocked, MCM helicase may decouple and unwind DNA ahead of the fork, exposing hundreds of bases of single-stranded DNA (ssDNA)
[40][41]. As with ssDNA exposed by 5′–3′ resection of broken ends at DSBs by resection nucleases EXO1 and DNA2 (with its cofactor BLM)
[42][43][44], the ssDNA exposed by decoupled MCM helicase is rapidly bound by the abundant, heterotrimeric replication protein A (RPA) (
Figure 1A). RPA-bound ssDNA is recognized by the ATR (ataxia telangiectasia and Rad3-related) cofactor ATRIP (ATR-interacting protein), leading to activation of ATR (
Figure 1B), the central signaling kinase of the intra-S checkpoint response
[45]. In addition to ssDNA-RPA and ATRIP, ATR activation requires several other factors including TopBP1, RAD17-RFC, and the 9-1-1 complex. Additional, distinct ATR activation mechanisms have been described involving NBS1, a component of the MRE11-RAD50-NBS1 (MRN) complex, and the RPA-binding factor ETAA1
[45][46][47].
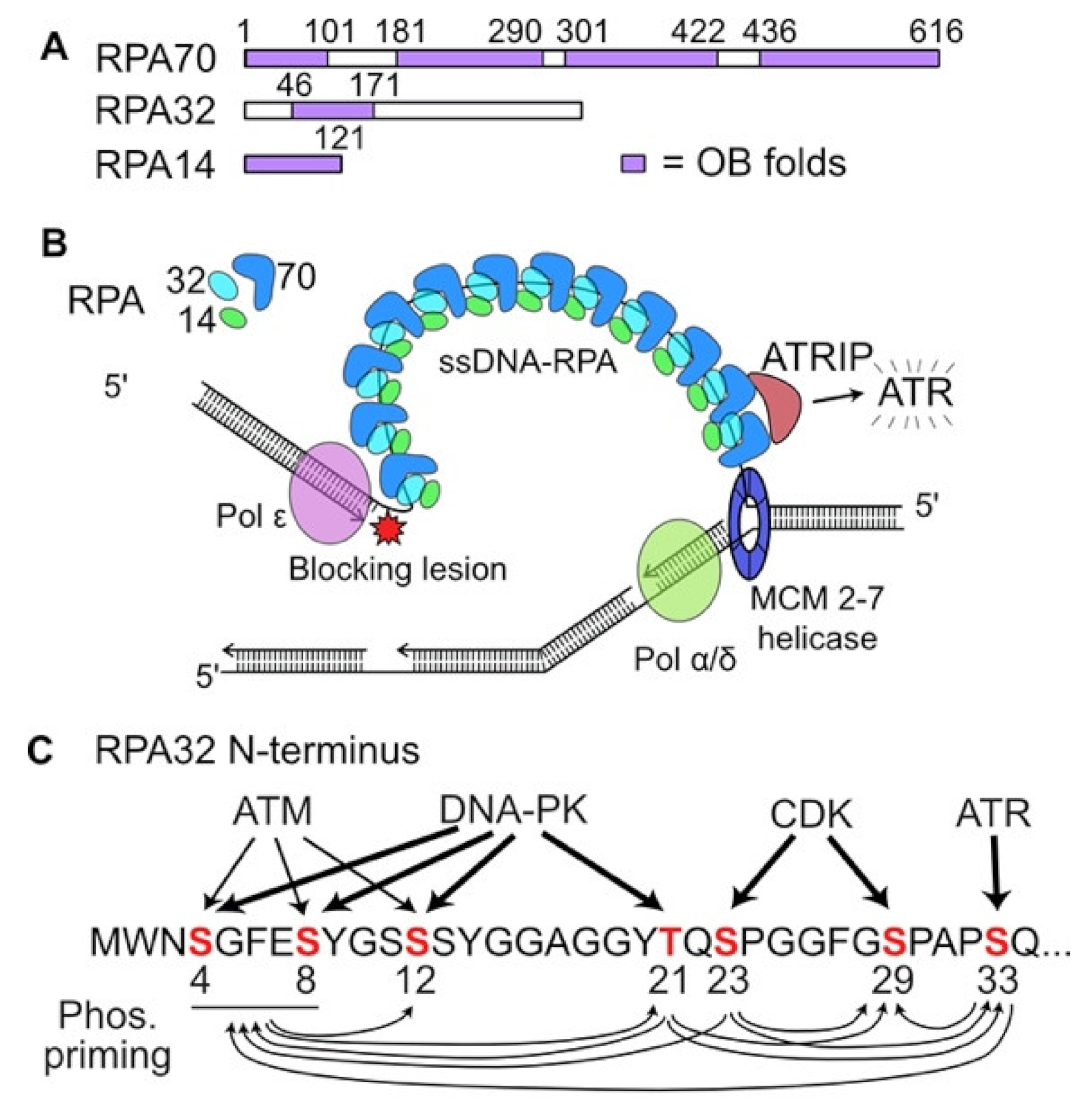
ATR is a member of the phosphatidyl inositol 3′ kinase-related kinase (PIKK) family, which also includes ATM and the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs). PIKKs play central roles in DNA damage responses including DSB repair, checkpoint activation, apoptosis, suppression of transcription, and responses to telomere dysfunction and viral infection
[48][49]. Activation of each PIKK involves a specific co-factor. The ATR interacting protein ATRIP recruits ATR to RPA-bound ssDNA, initiating ATR activation
[50]. The MRN complex and the Ku70/Ku80 heterodimer bind to frank DSB ends, the NBS1 component of MRN recruits and activates ATM, and Ku70/Ku80 recruits and activates DNA-PKcs
[48]. Activated PIKKs are autophosphorylated, and they phosphorylate each other and many other targets, showing various degrees of signaling pathway crosstalk
[48].
3. Protecting and Rescuing Blocked Replication Forks
DNA replication initiates at origins in a complex, highly regulated process involving assembly of pre-replication complexes and licensing factors that ensure DNA is replicated only once per cell cycle
[51]. For this reason, there is a premium on protecting replisomes at stalled replication forks to prevent replisome dissociation and fork collapse. Stalled forks are protected by a plethora of repair and replication factors, including RIF1, which inhibits end resection, the MRN-interacting protein MRNIP, the TLS suppressor USP1 which regulates PCNA via de-ubiquitination, HR proteins (RAD51, BRCA1, BRCA2, FANCD2), and RADX which regulates RAD51
[30][52][53][54][55][56][57]. Cells with defects in any of these fork protection factors are hypersensitive to replication stress.
Maintaining replisomes to protect stalled forks often involves fork regression to a ‘chicken foot’ structure that resembles four-way branched Holliday junctions of HR reactions (
Figure 2A)
[29]. Chicken foot structures have a one-ended DSB that at least initially includes ssDNA to which the HR factors RAD51, BRCA1, BRCA2, and the RAD51 paralogs (RAD51B/C/D and XRCC2/3) are recruited
[58], although HR factors appear to play distinct roles in HR and fork protection
[59].
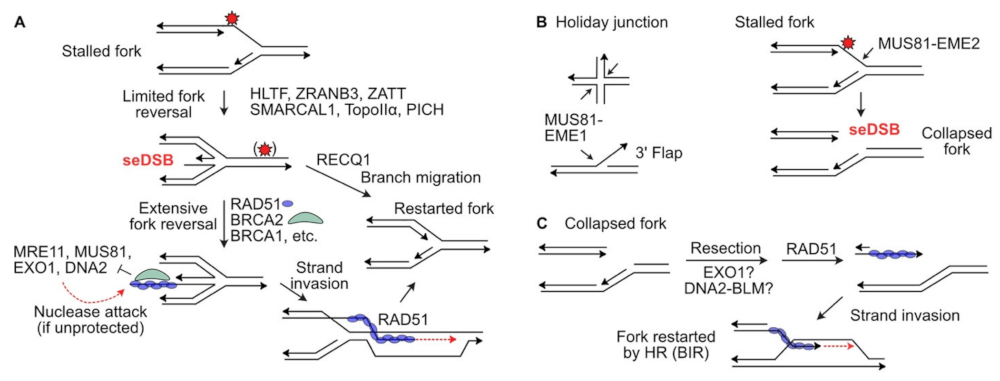
4. MUS81: An Ancient Structure-Specific Nuclease Involved in HR and Restart of Stressed Replication Forks
MUS81 is a structure-specific 3′ endonuclease in the XPF 3′ endonuclease family that cleaves a variety of branched DNA structures including 3′ flaps and Holliday junctions. Yeast Mus81 was first discovered in 2000 in a two-hybrid screen for proteins that interacted with the RAD54 HR protein and was named for the sensitivity of Mus81-defective cells to methyl methanesulfonate and UV light
[60]. Mus81-defective yeast also have a severe meiotic HR defect that together with its interaction with RAD54 suggested an important role in HR
[60]. Indeed, yeast Mus81 and its Eme1 cofactor resolve Holliday junctions and human MUS81 cleaves four-way (Holliday) junctions and 3′ flap structures
[61][62] (
Figure 2B). In human cells, MUS81 with its EME1 cofactor resolves Holliday junctions in HR intermediates
[63][64][65][66], and reversed forks that resemble Holliday junctions
[67]. In contrast, MUS81 with its EME2 cofactor cleaves blocked replication forks, causing fork collapse to a seDSB
[68][69][70] (
Figure 2B). The seDSB is apparently resected to allow formation of a RAD51-ssDNA nucleoprotein filament that catalyzes fork restart by a mechanism that resembles break-induced replication (BIR)
[71], although the resection nuclease(s) involved in processing MUS81-cleaved forks are not known (
Figure 2C). Yeast Mus81 also mediates resolution of structures in G2/M that arise when blocked forks are rescued by converging forks to complete DNA replication
[72].
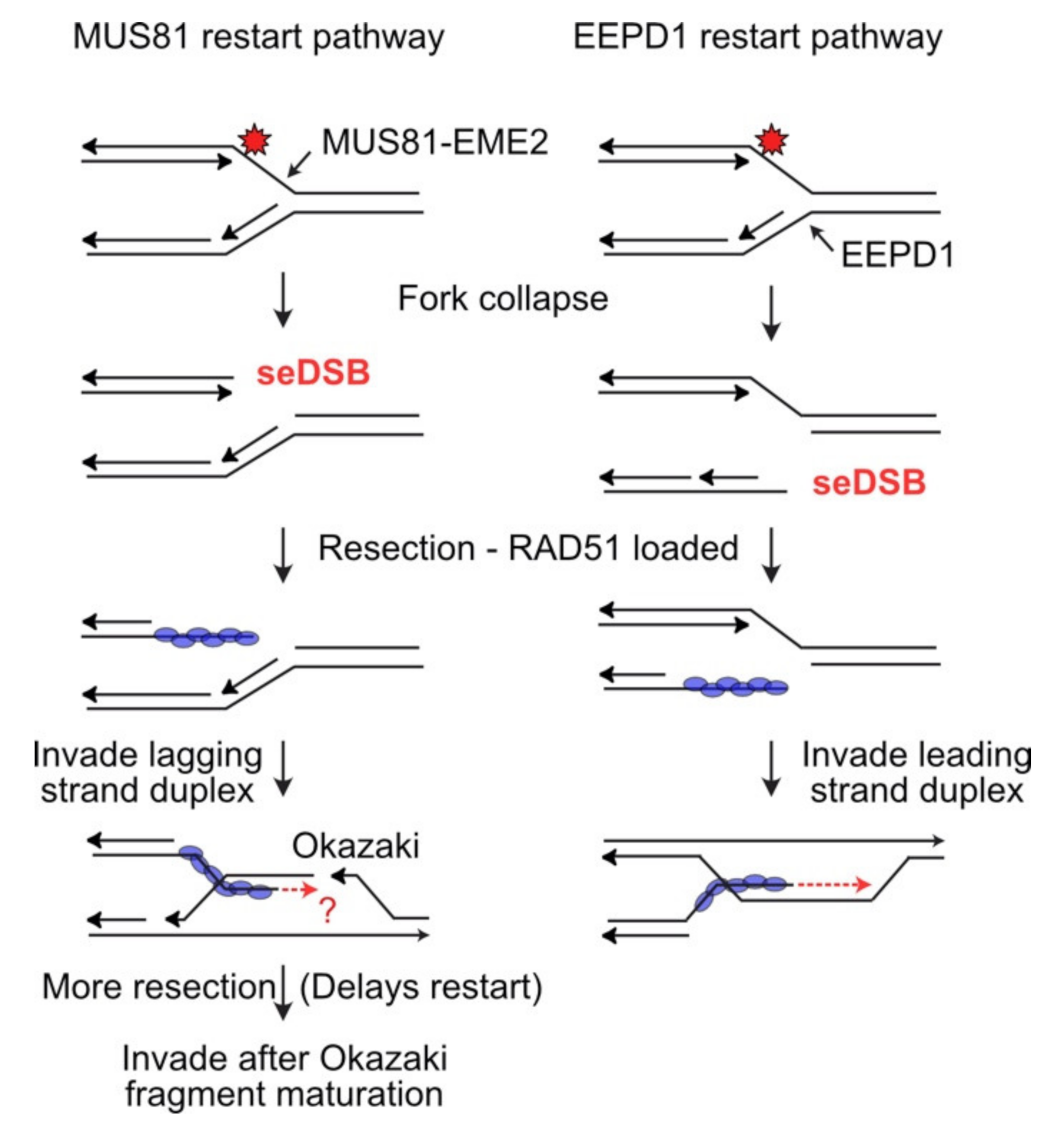
5. EEPD1: A 5′ Structure-Specific Endonuclease That Complements the 3′ MUS81 Nuclease
EEPD1 (endo- exonuclease phosphatase domain protein 1) has a DNase I-like nuclease domain and a DNA binding domain with two helix–hairpin–helix motifs similar to those in prokaryotic RuvA2. As with MUS81, cells with defective EEPD1 are hypersensitive to a variety of genotoxic chemicals and radiation, and replication stress induces chromosome aberrations and mitotic catastrophe
[73][74]. iPOND (isolation of proteins on nascent DNA) is a technique that reveals proteins associated with replication forks, including replisome components and proteins recruited to stressed forks
[75][76]. iPOND analysis demonstrated that EEPD1 is recruited to stalled replication forks, and similar to MUS81, EEPD1 cleaves fork structures in vitro, and stalled replication forks in vivo (
Figure 3)
[73].
6. Metnase: A Recently Evolved Nuclease-Protein Methyl Transferase That Promotes Replication Fork Restart
Metnase evolved ~50 million years ago when a
Mariner transposon integrated downstream of a SET protein methylase, and subsequent genetic changes fused the SET and nuclease domains
[77]. Metnase is a structure-specific nuclease with numerous genome stabilization functions including promotion of cNHEJ, chromosome decatenation, and restart of stressed replication forks
[78][79][80]. Although defects in the Metnase nuclease delay replication fork restart
[81] and Metnase cleaves replication fork structures in vitro
[82], Metnase does not cleave stalled forks in vivo like MUS81 and EEPD1
[74]. These findings suggest that Metnase nuclease functions in a later step in replication fork restart, such as trimming flaps in HR-mediated fork repair intermediates
[74]. The Metnase protein methylase targets histone H3 K36 to promote recruitment of cNHEJ factors Ku and NBS1
[83], Metnase automethylation regulates its chromosome decatenation function
[84], and its methylase also plays an as yet undefined role in promoting restart of stressed replication forks
[85]. Metnase is phosphorylated by Chk1, and this modification promotes cNHEJ, but suppresses replication fork restart
[86]. Indeed, Metnase regulates Chk1 stability, suggesting a feedback loop between Metnase and Chk1 that coordinates DNA repair and checkpoint processes
[87].
7. Conclusions
Despite the major advances in molecular characterization of tumors that inform targeted cancer therapies, the majority of cancer patients still receive non-targeted, genotoxic chemo- or radiotherapy, and these genotoxins universally cause replication stress. This has stimulated drug development efforts to augment chemo- and radiotherapy with agents that block general DDR signaling, such as inhibitors of ATM and ATR
[88][89][90], as well as HR factors and replication stress nucleases (
Table 1). Characterizing expression levels of MUS81, EEPD1, and Metnase may help illuminate tumor resistance to traditional therapeutics and inform personalized treatments, such as higher genotoxin doses to counteract the enhanced replication stress resistance associated with overexpression of these nucleases. Because of the central nature of DNA replication in cell division, replication stress responses also provide a rich environment for the development of targeted, synthetic lethal treatment strategies
[34].
Table 1. Functions and inhibitors of key replication stress nucleases and co-factors.
| Protein |
Biochemical Activities |
Biological Functions |
Inhibitor References |
| RPA |
Binds ssDNA, ATRIP, and itself |
DNA replication and repair; activates ATR through ATRIP binding to RPA-bound ssDNA; replaced by RAD51 on ssDNA during HR |
[91][92][93][94][95] |
| MRE11 |
DSB end binding, 3′-5′ exonuclease, endonuclease |
Early DSB sensor, ATM activation, promotes cNHEJ, initiates resection for HR |
[96][97][98] |
| CtIP |
Endonuclease |
Promotes limited resection by MRE11 |
[99][100] |
| EXO1 |
5′-3′ exonuclease |
Extensive end resection |
[101] * |
| DNA2 |
5′-3′ exonuclease |
Extensive end resection |
[102][103] |
| BLM |
3′-5′ helicase |
Unwinds DNA structures during HR, promotes resection by DNA2 |
[104][105][106][107] |
| RAD51 |
Strand invasion (recombinase) |
Binds dsDNA, ssDNA and itself, catalyzes HR |
[108][109][110][111][112][113] |
| MUS81-EME2 |
3′ structure specific endonuclease |
Cleaves stalled forks, promotes fork restart |
[114] |
| EEPD1 |
5′ structure specific endonuclease |
Cleaves stalled forks, promotes fork restart and fork resection by EXO1 |
None † |
| Metnase |
5′ structure specific endonuclease, protein methylase |
Cleaves stalled forks, promotes fork restart and fork resection by EXO1 |
[115] |
| SLX1-SLX4 |
5′ structure specific endonuclease |
Cleaves branched structures, promotes HR, crosslink repair, and telomere maintenance |
None † |
| XPF-ERCC1 |
5′ structure specific endonuclease |
Nucleotide excision repair, inter-strand crosslink repair, HR (replication stress?) |
[116][117] |
 +1 credit
+1 credit

