 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Tuyet Thi Nguyen | + 4906 word(s) | 4906 | 2022-03-07 09:04:17 | | | |
| 2 | Nhung Thi Nguyen | Meta information modification | 4906 | 2022-03-24 13:57:47 | | | | |
| 3 | Kyu-Sang Park | Meta information modification | 4906 | 2022-03-25 01:05:05 | | | | |
| 4 | Bruce Ren | + 113 word(s) | 5019 | 2022-03-25 04:09:42 | | |
Video Upload Options
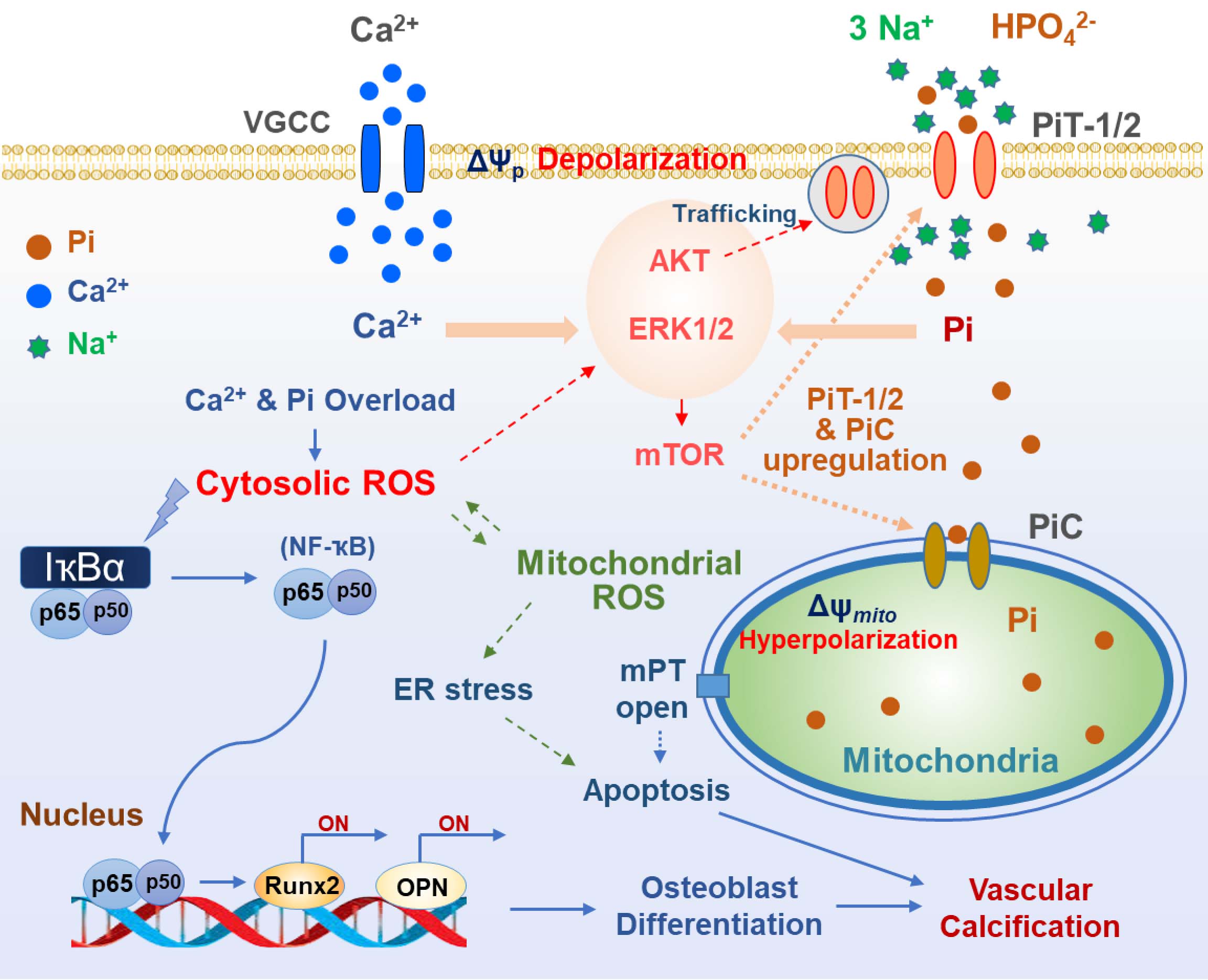
Inorganic phosphate (Pi) is essential for maintaining cellular function but excess of Pi leads to serious complications, including vascular calcification. Accumulating evidence suggests that oxidative stress contributes to the pathogenic progression of calcific changes. However, the molecular mechanism underlying Pi-induced reactive oxygen species (ROS) generation and its detrimental consequences remain unclear. Type III Na+-dependent Pi cotransporter, PiT-1/-2, play a significant role in Pi uptake of vascular smooth muscle cells. Pi influx via PiT-1/-2 increases the abundance of PiT-1/-2 and depolarization-activated Ca2+ entry due to its electrogenic properties, which may lead to Ca2+ and Pi overload and oxidative stress. At least four mitochondrial Pi transporters are suggested, among which the phosphate carrier (PiC) is known to be mainly involved in mitochondrial Pi uptake. Pi transport via PiC may induce hyperpolarization and superoxide generation, which may lead to mitochondrial dysfunction and endoplasmic reticulum stress, together with generation of cytosolic ROS. Increase in net influx of Ca2+ and Pi and their accumulation in the cytosol and mitochondrial matrix synergistically increases oxidative stress and osteogenic differentiation, which could be prevented by suppressing either Ca2+ or Pi overload. Therapeutic strategies targeting plasmalemmal and mitochondrial Pi transports can protect against Pi-induced oxidative stress and vascular calcification.
1. Introduction
2. Plasmalemmal Phosphate Transporters
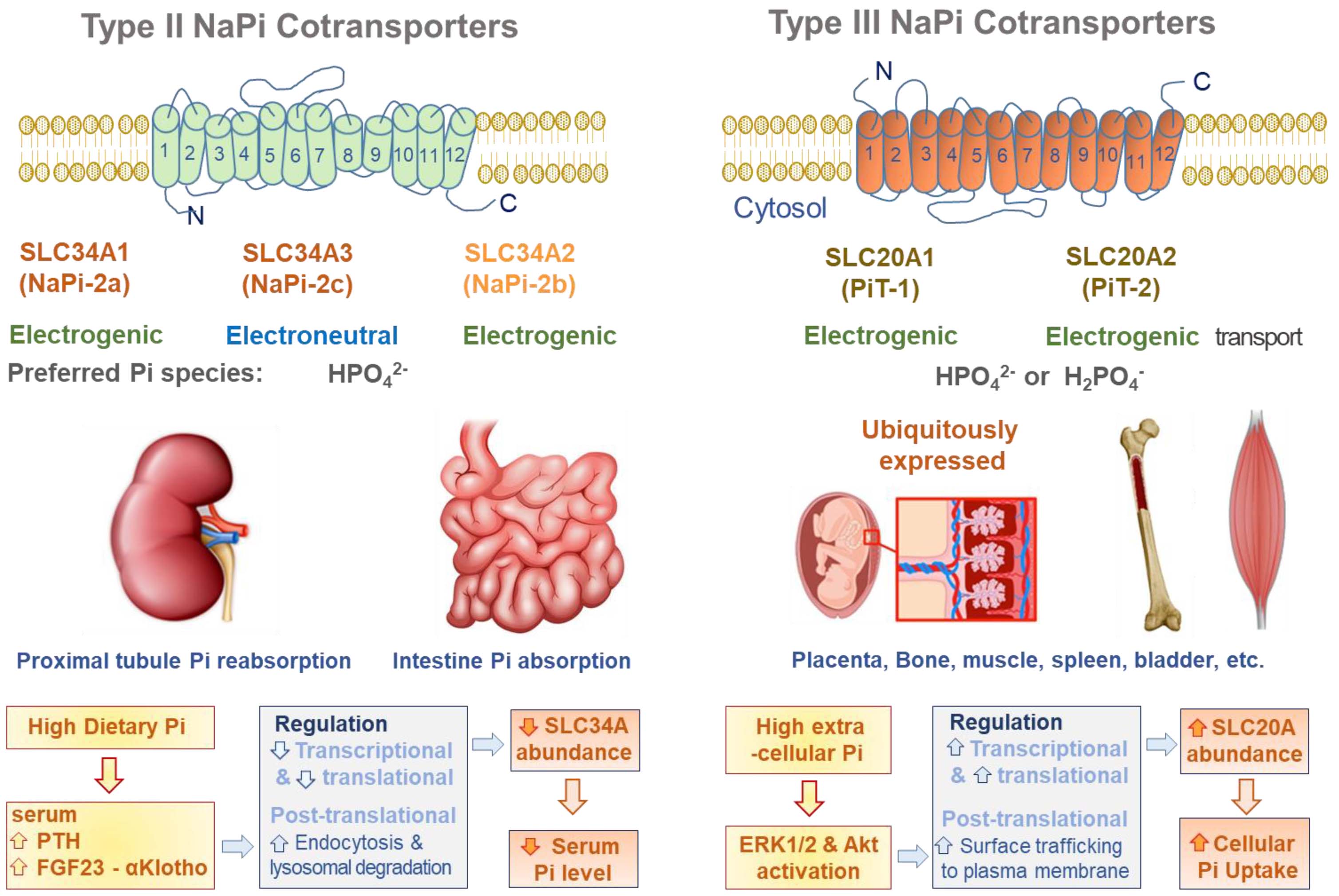
2.1. SLC34A Family
2.2. SLC20A Family
2.3. Cytosolic Phosphate Exporter
2.4. Oxidative Stress Related to Plasmalemmal Pi Transporters
3. Mitochondrial Phosphate Transporters
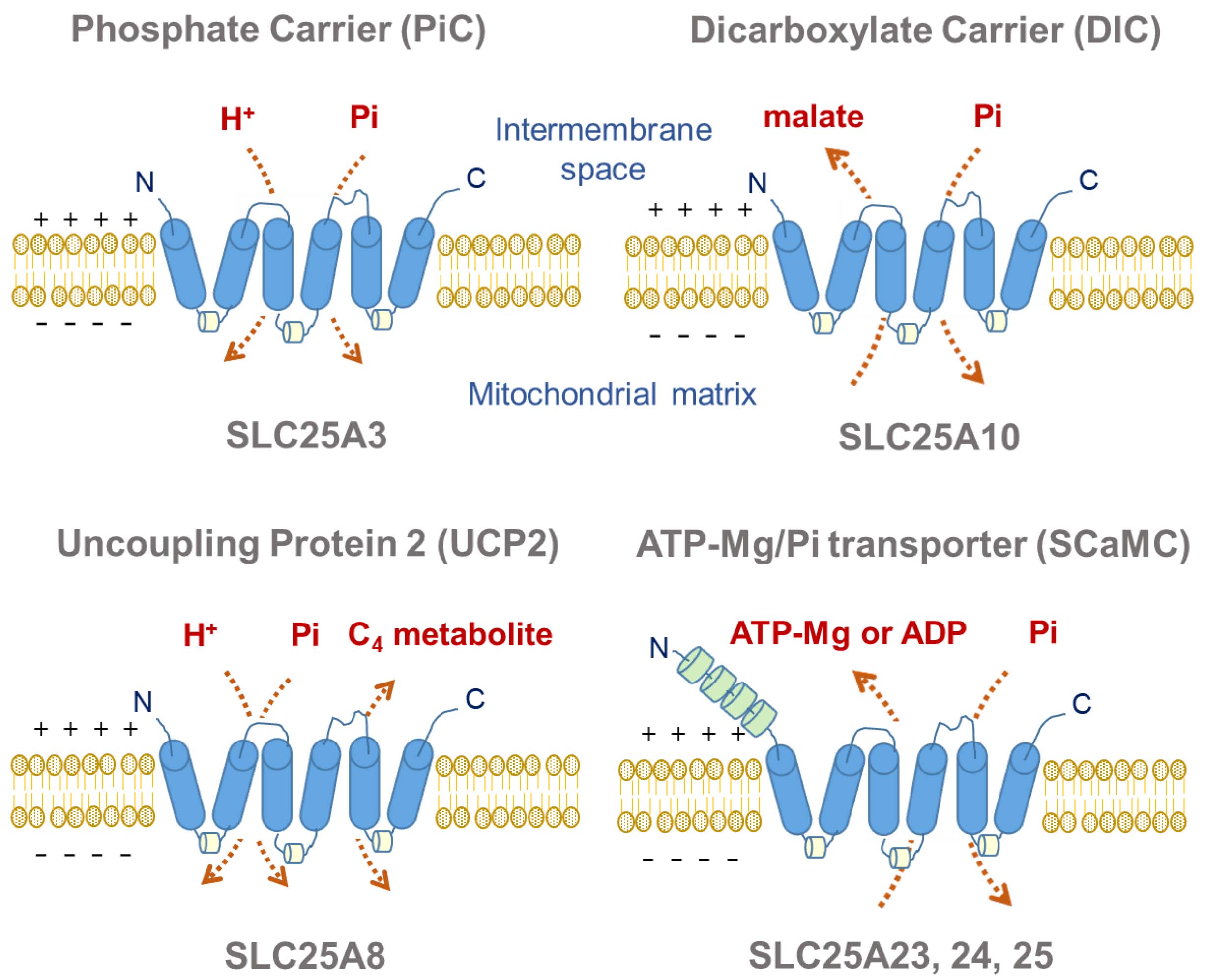
3.1. PiC
3.2. DIC
3.3. UCP2
3.4. Mg-ATP/Pi Carrier
3.5. Oxidative Stress Due to Mitochondrial Pi Transport
References
- Yamamoto, Y.; Ishikawa, Y.; Shimpo, M.; Matsumura, M. Mönckeberg’s sclerosis. J. Gen. Fam. Med. 2021, 22, 55–56.
- Lee, S.J.; Lee, I.K.; Jeon, J.H. Vascular calcification-new insights into its mechanism. Int. J. Mol. Sci. 2020, 21, 2685.
- Ho, C.Y.; Shanahan, C.M. Medial arterial calcification: An overlooked player in peripheral arterial disease. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1475–1482.
- Quan, X.; Das, R.; Xu, S.; Cline, G.W.; Wiederkehr, A.; Wollheim, C.B.; Park, K.S. Mitochondrial phosphate transport during nutrient stimulation of ins-1e insulinoma cells. Mol. Cell. Endocrinol. 2013, 381, 198–209.
- Biber, J.; Hernando, N.; Forster, I. Phosphate transporters and their function. Annu. Rev. Physiol. 2013, 75, 535–550.
- Forster, I.C.; Hernando, N.; Biber, J.; Murer, H. Phosphate transporters of the slc20 and slc34 families. Mol. Asp. Med. 2013, 34, 386–395.
- Wagner, C.A.; Hernando, N.; Forster, I.C.; Biber, J. The slc34 family of sodium-dependent phosphate transporters. Pflugers Arch. 2014, 466, 139–153.
- Forster, I.C.; Hernando, N.; Biber, J.; Murer, H. Phosphate transport kinetics and structure-function relationships of slc34 and slc20 proteins. Curr. Top. Membr. 2012, 70, 313–356.
- Miyamoto, K.; Haito-Sugino, S.; Kuwahara, S.; Ohi, A.; Nomura, K.; Ito, M.; Kuwahata, M.; Kido, S.; Tatsumi, S.; Kaneko, I.; et al. Sodium-dependent phosphate cotransporters: Lessons from gene knockout and mutation studies. J. Pharm. Sci. 2011, 100, 3719–3730.
- Breusegem, S.Y.; Takahashi, H.; Giral-Arnal, H.; Wang, X.; Jiang, T.; Verlander, J.W.; Wilson, P.; Miyazaki-Anzai, S.; Sutherland, E.; Caldas, Y.; et al. Differential regulation of the renal sodium-phosphate cotransporters napi-iia, napi-iic, and pit-2 in dietary potassium deficiency. Am. J. Physiol. Renal Physiol. 2009, 297, F350–F361.
- Ghezzi, C.; Meinild, A.K.; Murer, H.; Forster, I.C. Voltage- and substrate-dependent interactions between sites in putative re-entrant domains of a Na(+)-coupled phosphate cotransporter. Pflugers Arch. 2011, 461, 645–663.
- Moschèn, I.; Setiawan, I.; Bröer, S.; Murer, H.; Lang, F. Effect of napi-mediated phosphate transport on intracellular ph. Pflugers Arch. 2001, 441, 802–806.
- Segawa, H.; Aranami, F.; Kaneko, I.; Tomoe, Y.; Miyamoto, K. The roles of na/pi-ii transporters in phosphate metabolism. Bone 2009, 45 (Suppl. S1), S2–S7.
- Segawa, H.; Onitsuka, A.; Furutani, J.; Kaneko, I.; Aranami, F.; Matsumoto, N.; Tomoe, Y.; Kuwahata, M.; Ito, M.; Matsumoto, M.; et al. Npt2a and npt2c in mice play distinct and synergistic roles in inorganic phosphate metabolism and skeletal development. Am. J. Physiol. Renal Physiol. 2009, 297, F671–F678.
- Li, X.; Yang, H.Y.; Giachelli, C.M. Role of the sodium-dependent phosphate cotransporter, pit-1, in vascular smooth muscle cell calcification. Circ. Res. 2006, 98, 905–912.
- Villa-Bellosta, R.; Bogaert, Y.E.; Levi, M.; Sorribas, V. Characterization of phosphate transport in rat vascular smooth muscle cells: Implications for vascular calcification. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1030–1036.
- Voelkl, J.; Alesutan, I.; Leibrock, C.B.; Quintanilla-Martinez, L.; Kuhn, V.; Feger, M.; Mia, S.; Ahmed, M.S.; Rosenblatt, K.P.; Kuro, O.M.; et al. Spironolactone ameliorates pit1-dependent vascular osteoinduction in klotho-hypomorphic mice. J. Clin. Investig. 2013, 123, 812–822.
- Sekiguchi, S.; Suzuki, A.; Asano, S.; Nishiwaki-Yasuda, K.; Shibata, M.; Nagao, S.; Yamamoto, N.; Matsuyama, M.; Sato, Y.; Yan, K.; et al. Phosphate overload induces podocyte injury via type iii na-dependent phosphate transporter. Am. J. Physiol. Renal Physiol. 2011, 300, F848–F856.
- Werner, A.; Dehmelt, L.; Nalbant, P. Na+-dependent phosphate cotransporters: The napi protein families. J. Exp. Biol. 1998, 201, 3135–3142.
- Bergwitz, C.; Roslin, N.M.; Tieder, M.; Loredo-Osti, J.C.; Bastepe, M.; Abu-Zahra, H.; Frappier, D.; Burkett, K.; Carpenter, T.O.; Anderson, D.; et al. Slc34a3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter napi-iic in maintaining phosphate homeostasis. Am. J. Hum. Genet. 2006, 78, 179–192.
- Gonzalez-Parra, E.; Tuñón, J.; Egido, J.; Ortiz, A. Phosphate: A stealthier killer than previously thought? Cardiovasc. Pathol. 2012, 21, 372–381.
- Ohi, A.; Hanabusa, E.; Ueda, O.; Segawa, H.; Horiba, N.; Kaneko, I.; Kuwahara, S.; Mukai, T.; Sasaki, S.; Tominaga, R.; et al. Inorganic phosphate homeostasis in sodium-dependent phosphate cotransporter npt2b⁺/⁻ mice. Am. J. Physiol. Renal Physiol. 2011, 301, F1105–F1113.
- Miller, D.G.; Edwards, R.H.; Miller, A.D. Cloning of the cellular receptor for amphotropic murine retroviruses reveals homology to that for gibbon ape leukemia virus. Proc. Natl. Acad. Sci. USA 1994, 91, 78–82.
- O’Hara, B.; Johann, S.V.; Klinger, H.P.; Blair, D.G.; Rubinson, H.; Dunn, K.J.; Sass, P.; Vitek, S.M.; Robins, T. Characterization of a human gene conferring sensitivity to infection by gibbon ape leukemia virus. Cell Growth Differ. 1990, 1, 119–127.
- Kavanaugh, M.P.; Miller, D.G.; Zhang, W.; Law, W.; Kozak, S.L.; Kabat, D.; Miller, A.D. Cell-surface receptors for gibbon ape leukemia virus and amphotropic murine retrovirus are inducible sodium-dependent phosphate symporters. Proc. Natl. Acad. Sci. USA 1994, 91, 7071–7075.
- Olah, Z.; Lehel, C.; Anderson, W.B.; Eiden, M.V.; Wilson, C.A. The cellular receptor for gibbon ape leukemia virus is a novel high affinity sodium-dependent phosphate transporter. J. Biol. Chem. 1994, 269, 25426–25431.
- Nguyen, T.T.; Quan, X.; Xu, S.; Das, R.; Cha, S.K.; Kong, I.D.; Shong, M.; Wollheim, C.B.; Park, K.S. Intracellular alkalinization by phosphate uptake via type iii sodium-phosphate cotransporter participates in high-phosphate-induced mitochondrial oxidative stress and defective insulin secretion. FASEB J. 2016, 30, 3979–3988.
- Beck, L.; Leroy, C.; Beck-Cormier, S.; Forand, A.; Salaün, C.; Paris, N.; Bernier, A.; Ureña-Torres, P.; Prié, D.; Ollero, M.; et al. The phosphate transporter pit1 (slc20a1) revealed as a new essential gene for mouse liver development. PLoS ONE 2010, 5, e9148.
- Jensen, N.; Schrøder, H.D.; Hejbøl, E.K.; Füchtbauer, E.M.; de Oliveira, J.R.; Pedersen, L. Loss of function of slc20a2 associated with familial idiopathic basal ganglia calcification in humans causes brain calcifications in mice. J. Mol. Neurosci. 2013, 51, 994–999.
- Ren, Y.; Shen, Y.; Si, N.; Fan, S.; Zhang, Y.; Xu, W.; Shi, L.; Zhang, X. Slc20a2-deficient mice exhibit multisystem abnormalities and impaired spatial learning memory and sensorimotor gating but normal motor coordination abilities. Front. Genet. 2021, 12, 639935.
- Crouthamel, M.H.; Lau, W.L.; Leaf, E.M.; Chavkin, N.W.; Wallingford, M.C.; Peterson, D.F.; Li, X.; Liu, Y.; Chin, M.T.; Levi, M.; et al. Sodium-dependent phosphate cotransporters and phosphate-induced calcification of vascular smooth muscle cells: Redundant roles for pit-1 and pit-2. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2625–2632.
- Nguyen, N.T.; Nguyen, T.T.; Ly, D.D.; Xia, J.-B.; Qi, X.-F.; Lee, I.-K.; Cha, S.-K.; Park, K.-S. Oxidative stress by Ca2+ overload is critical for phosphate-induced vascular calcification. Am. J. Physiol. -Heart Circ. Physiol. 2020, 319, H1302–H1312.
- Yamada, S.; Leaf, E.M.; Chia, J.J.; Cox, T.C.; Speer, M.Y.; Giachelli, C.M. Pit-2, a type iii sodium-dependent phosphate transporter, protects against vascular calcification in mice with chronic kidney disease fed a high-phosphate diet. Kidney Int. 2018, 94, 716–727.
- Chen, W.C.; Li, Q.L.; Pan, Q.; Zhang, H.Y.; Fu, X.Y.; Yao, F.; Wang, J.N.; Yang, A.K. Xenotropic and polytropic retrovirus receptor 1 (xpr1) promotes progression of tongue squamous cell carcinoma (tscc) via activation of nf-κb signaling. J. Exp. Clin. Cancer Res. 2019, 38, 167.
- Secco, D.; Wang, C.; Arpat, B.A.; Wang, Z.; Poirier, Y.; Tyerman, S.D.; Wu, P.; Shou, H.; Whelan, J. The emerging importance of the spx domain-containing proteins in phosphate homeostasis. New Phytol. 2012, 193, 842–851.
- Legati, A.; Giovannini, D.; Nicolas, G.; López-Sánchez, U.; Quintáns, B.; Oliveira, J.R.; Sears, R.L.; Ramos, E.M.; Spiteri, E.; Sobrido, M.J.; et al. Mutations in xpr1 cause primary familial brain calcification associated with altered phosphate export. Nat. Genet. 2015, 47, 579–581.
- Ansermet, C.; Moor, M.B.; Centeno, G.; Auberson, M.; Hu, D.Z.; Baron, R.; Nikolaeva, S.; Haenzi, B.; Katanaeva, N.; Gautschi, I.; et al. Renal fanconi syndrome and hypophosphatemic rickets in the absence of xenotropic and polytropic retroviral receptor in the nephron. J. Am. Soc. Nephrol. 2017, 28, 1073–1078.
- Krämer, R. Structural and functional aspects of the phosphate carrier from mitochondria. Kidney Int. 1996, 49, 947–952.
- Palmieri, L.; Palmieri, F.; Runswick, M.J.; Walker, J.E. Identification by bacterial expression and functional reconstitution of the yeast genomic sequence encoding the mitochondrial dicarboxylate carrier protein. FEBS Lett. 1996, 399, 299–302.
- Fiermonte, G.; De Leonardis, F.; Todisco, S.; Palmieri, L.; Lasorsa, F.M.; Palmieri, F. Identification of the mitochondrial atp-mg/pi transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution. J. Biol. Chem. 2004, 279, 30722–30730.
- Vozza, A.; Parisi, G.; De Leonardis, F.; Lasorsa, F.M.; Castegna, A.; Amorese, D.; Marmo, R.; Calcagnile, V.M.; Palmieri, L.; Ricquier, D.; et al. Ucp2 transports c4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. USA 2014, 111, 960–965.
- Palmieri, F. The mitochondrial transporter family slc25: Identification, properties and physiopathology. Mol. Asp. Med. 2013, 34, 465–484.
- Runswick, M.J.; Powell, S.J.; Nyren, P.; Walker, J.E. Sequence of the bovine mitochondrial phosphate carrier protein: Structural relationship to adp/atp translocase and the brown fat mitochondria uncoupling protein. EMBO J. 1987, 6, 1367–1373.
- Dolce, V.; Fiermonte, G.; Palmieri, F. Tissue-specific expression of the two isoforms of the mitochondrial phosphate carrier in bovine tissues. FEBS Lett. 1996, 399, 95–98.
- Palmieri, F.; Bisaccia, F.; Capobianco, L.; Dolce, V.; Fiermonte, G.; Iacobazzi, V.; Zara, V. Transmembrane topology, genes, and biogenesis of the mitochondrial phosphate and oxoglutarate carriers. J. Bioenerg. Biomembr. 1993, 25, 493–501.
- Dolce, V.; Iacobazzi, V.; Palmieri, F.; Walker, J.E. The sequences of human and bovine genes of the phosphate carrier from mitochondria contain evidence of alternatively spliced forms. J. Biol. Chem. 1994, 269, 10451–10460.
- Boulet, A.; Vest, K.E.; Maynard, M.K.; Gammon, M.G.; Russell, A.C.; Mathews, A.T.; Cole, S.E.; Zhu, X.; Phillips, C.B.; Kwong, J.Q.; et al. The mammalian phosphate carrier slc25a3 is a mitochondrial copper transporter required for cytochrome c oxidase biogenesis. J. Biol. Chem. 2018, 293, 1887–1896.
- Fonyó, A.; Ligeti, E. The role of intramitochondrial pi in stimulation of respiration by calcium and strontium. FEBS Lett. 1978, 93, 289–292.
- Gutiérrez-Aguilar, M.; Douglas, D.L.; Gibson, A.K.; Domeier, T.L.; Molkentin, J.D.; Baines, C.P. Genetic manipulation of the cardiac mitochondrial phosphate carrier does not affect permeability transition. J. Mol. Cell. Cardiol. 2014, 72, 316–325.
- Kwong, J.; Davis, J.; Baines, C.; Sargent, M.; Karch, J.; Wang, X.; Huang, T.; Molkentin, J. Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death Differ. 2014, 21, 1209–1217.
- Seifert, E.L.; Ligeti, E.; Mayr, J.A.; Sondheimer, N.; Hajnóczky, G. The mitochondrial phosphate carrier: Role in oxidative metabolism, calcium handling and mitochondrial disease. Biochem. Biophys. Res. Commun. 2015, 464, 369–375.
- Palmieri, F.; Scarcia, P.; Monné, M. Diseases caused by mutations in mitochondrial carrier genes slc25: A review. Biomolecules 2020, 10, 655.
- Mayr, J.A.; Merkel, O.; Kohlwein, S.D.; Gebhardt, B.R.; Böhles, H.; Fötschl, U.; Koch, J.; Jaksch, M.; Lochmüller, H.; Horváth, R.; et al. Mitochondrial phosphate-carrier deficiency: A novel disorder of oxidative phosphorylation. Am. J. Hum. Genet. 2007, 80, 478–484.
- Mayr, J.A.; Zimmermann, F.A.; Horváth, R.; Schneider, H.-C.; Schoser, B.; Holinski-Feder, E.; Czermin, B.; Freisinger, P.; Sperl, W. Deficiency of the mitochondrial phosphate carrier presenting as myopathy and cardiomyopathy in a family with three affected children. Neuromuscul. Disord. 2011, 21, 803–808.
- Zorov, D.B.; Juhaszova, M.; Yaniv, Y.; Nuss, H.B.; Wang, S.; Sollott, S.J. Regulation and pharmacology of the mitochondrial permeability transition pore. Cardiovasc. Res. 2009, 83, 213–225.
- Leung, A.W.; Varanyuwatana, P.; Halestrap, A.P. The mitochondrial phosphate carrier interacts with cyclophilin d and may play a key role in the permeability transition. J. Biol. Chem. 2008, 283, 26312–26323.
- Varanyuwatana, P.; Halestrap, A.P. The roles of phosphate and the phosphate carrier in the mitochondrial permeability transition pore. Mitochondrion 2012, 12, 120–125.
- Basso, E.; Petronilli, V.; Forte, M.A.; Bernardi, P. Phosphate is essential for inhibition of the mitochondrial permeability transition pore by cyclosporin a and by cyclophilin d ablation. J. Biol. Chem. 2008, 283, 26307–26311.
- Poncet, D.; Pauleau, A.L.; Szabadkai, G.; Vozza, A.; Scholz, S.R.; Le Bras, M.; Brière, J.J.; Jalil, A.; Le Moigne, R.; Brenner, C.; et al. Cytopathic effects of the cytomegalovirus-encoded apoptosis inhibitory protein vmia. J. Cell Biol. 2006, 174, 985–996.
- Poncet, D.; Larochette, N.; Pauleau, A.L.; Boya, P.; Jalil, A.A.; Cartron, P.F.; Vallette, F.; Schnebelen, C.; Bartle, L.M.; Skaletskaya, A.; et al. An anti-apoptotic viral protein that recruits bax to mitochondria. J. Biol. Chem. 2004, 279, 22605–22614.
- Pauleau, A.L.; Galluzzi, L.; Scholz, S.R.; Larochette, N.; Kepp, O.; Kroemer, G. Unexpected role of the phosphate carrier in mitochondrial fragmentation. Cell Death Differ. 2008, 15, 616–618.
- Nishi, Y.; Fujimoto, S.; Sasaki, M.; Mukai, E.; Sato, H.; Sato, Y.; Tahara, Y.; Nakamura, Y.; Inagaki, N. Role of mitochondrial phosphate carrier in metabolism-secretion coupling in rat insulinoma cell line ins-1. Biochem. J. 2011, 435, 421–430.
- Palmieri, F. Mitochondrial carrier proteins. FEBS Lett. 1994, 346, 48–54.
- Das, K.; Lewis, R.Y.; Combatsiaris, T.P.; Lin, Y.; Shapiro, L.; Charron, M.J.; Scherer, P.E. Predominant expression of the mitochondrial dicarboxylate carrier in white adipose tissue. Biochem. J. 1999, 344 Pt 2, 313–320.
- Fiermonte, G.; Dolce, V.; Arrigoni, R.; Runswick, M.J.; Walker, J.E.; Palmieri, F. Organization and sequence of the gene for the human mitochondrial dicarboxylate carrier: Evolution of the carrier family. Biochem. J. 1999, 344 Pt 3, 953–960.
- Coty, W.A.; Pedersen, P.L. Phosphate transport in rat liver mitochondria. Kinetics and energy requirements. J. Biol. Chem. 1974, 249, 2593–2598.
- Hlouschek, J.; Ritter, V.; Wirsdörfer, F.; Klein, D.; Jendrossek, V.; Matschke, J. Targeting slc25a10 alleviates improved antioxidant capacity and associated radioresistance of cancer cells induced by chronic-cycling hypoxia. Cancer Lett. 2018, 439, 24–38.
- Johnson, R.N.; Chappell, J.B. The transport of inorganic phosphate by the mitochondrial dicarboxylate carrier. Biochem. J. 1973, 134, 769–774.
- Chen, Z.; Lash, L.H. Evidence for mitochondrial uptake of glutathione by dicarboxylate and 2-oxoglutarate carriers. J. Pharmacol. Exp. Ther. 1998, 285, 608–618.
- Kamga, C.K.; Zhang, S.X.; Wang, Y. Dicarboxylate carrier-mediated glutathione transport is essential for reactive oxygen species homeostasis and normal respiration in rat brain mitochondria. Am. J. Physiol. Cell Physiol. 2010, 299, C497–C505.
- Huypens, P.; Pillai, R.; Sheinin, T.; Schaefer, S.; Huang, M.; Odegaard, M.L.; Ronnebaum, S.M.; Wettig, S.D.; Joseph, J.W. The dicarboxylate carrier plays a role in mitochondrial malate transport and in the regulation of glucose-stimulated insulin secretion from rat pancreatic beta cells. Diabetologia 2011, 54, 135–145.
- Punzi, G.; Porcelli, V.; Ruggiu, M.; Hossain, M.F.; Menga, A.; Scarcia, P.; Castegna, A.; Gorgoglione, R.; Pierri, C.L.; Laera, L. Slc25a10 biallelic mutations in intractable epileptic encephalopathy with complex i deficiency. Hum. Mol. Genet. 2018, 27, 499–504.
- Enerbäck, S.; Jacobsson, A.; Simpson, E.M.; Guerra, C.; Yamashita, H.; Harper, M.E.; Kozak, L.P. Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature 1997, 387, 90–94.
- Zhang, C.-Y.; Baffy, G.; Perret, P.; Krauss, S.; Peroni, O.; Grujic, D.; Hagen, T.; Vidal-Puig, A.J.; Boss, O.; Kim, Y.-B.; et al. Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, β cell dysfunction, and type 2 diabetes. Cell 2001, 105, 745–755.
- Tian, X.Y.; Ma, S.; Tse, G.; Wong, W.T.; Huang, Y. Uncoupling protein 2 in cardiovascular health and disease. Front. Physiol. 2018, 9, 1060.
- Fleury, C.; Neverova, M.; Collins, S.; Raimbault, S.; Champigny, O.; Levi-Meyrueis, C.; Bouillaud, F.; Seldin, M.F.; Surwit, R.S.; Ricquier, D.; et al. Uncoupling protein-2: A novel gene linked to obesity and hyperinsulinemia. Nat. Genet. 1997, 15, 269–272.
- Langin, D.; Larrouy, D.; Barbe, P.; Millet, L.; Viguerie-Bascands, N.; Andreelli, F.; Laville, M.; Vidal, H. Uncoupling protein-2 (ucp2) and uncoupling protein-3 (ucp3) expression in adipose tissue and skeletal muscle in humans. Int. J. Obes. Relat. Metab. Disord. 1999, 23 (Suppl. S6), S64–S67.
- Rupprecht, A.; Bräuer, A.U.; Smorodchenko, A.; Goyn, J.; Hilse, K.E.; Shabalina, I.G.; Infante-Duarte, C.; Pohl, E.E. Quantification of uncoupling protein 2 reveals its main expression in immune cells and selective up-regulation during t-cell proliferation. PLoS ONE 2012, 7, e41406.
- Pecqueur, C.; Alves-Guerra, M.C.; Gelly, C.; Levi-Meyrueis, C.; Couplan, E.; Collins, S.; Ricquier, D.; Bouillaud, F.; Miroux, B. Uncoupling protein 2, in vivo distribution, induction upon oxidative stress, and evidence for translational regulation. J. Biol. Chem. 2001, 276, 8705–8712.
- Berardi, M.J.; Chou, J.J. Fatty acid flippase activity of ucp2 is essential for its proton transport in mitochondria. Cell. Metab. 2014, 20, 541–552.
- Echtay, K.S.; Roussel, D.; St-Pierre, J.; Jekabsons, M.B.; Cadenas, S.; Stuart, J.A.; Harper, J.A.; Roebuck, S.J.; Morrison, A.; Pickering, S.; et al. Superoxide activates mitochondrial uncoupling proteins. Nature 2002, 415, 96–99.
- Chen, W.; Luo, S.; Xie, P.; Hou, T.; Yu, T.; Fu, X. Overexpressed ucp2 regulates mitochondrial flashes and reverses lipopolysaccharide-induced cardiomyocytes injury. Am. J. Transl. Res. 2018, 10, 1347–1356.
- Jezek, P.; Holendova, B.; Garlid, K.D.; Jaburek, M. Mitochondrial uncoupling proteins: Subtle regulators of cellular redox signaling. Antioxid. Redox Signal. 2018, 29, 667–714.
- He, M.; Ma, Y.; Wang, R.; Zhang, J.; Jing, L.; Li, P.A. Deletion of mitochondrial uncoupling protein 2 exacerbates mitochondrial damage in mice subjected to cerebral ischemia and reperfusion injury under both normo- and hyperglycemic conditions. Int. J. Biol. Sci. 2020, 16, 2788–2802.
- del Arco, A.; Satrústegui, J. Identification of a novel human subfamily of mitochondrial carriers with calcium-binding domains. J. Biol. Chem. 2004, 279, 24701–24713.
- Satrústegui, J.; Pardo, B.; Arco, A.d. Mitochondrial transporters as novel targets for intracellular calcium signaling. Physiol. Rev. 2007, 87, 29–67.
- Aprille, J.R.; Austin, J. Regulation of the mitochondrial adenine nucleotide pool size. Arch. Biochem. Biophys. 1981, 212, 689–699.
- Ryu, J.; Ko, J.M.; Shin, C.-H. A 9-year-old korean girl with fontaine progeroid syndrome: A case report with further phenotypical delineation and description of clinical course during long-term follow-up. BMC Med. Genet. 2019, 20, 188.
- Writzl, K.; Maver, A.; Kovacic, L.; Martinez-Valero, P.; Contreras, L.; Satrustegui, J.; Castori, M.; Faivre, L.; Lapunzina, P.; van Kuilenburg, A.B.P.; et al. De novo mutations in slc25a24 cause a disorder characterized by early aging, bone dysplasia, characteristic face, and early demise. Am. J. Hum. Genet. 2017, 101, 844–855.
- Ehmke, N.; Graul-Neumann, L.; Smorag, L.; Koenig, R.; Segebrecht, L.; Magoulas, P.; Scaglia, F.; Kilic, E.; Hennig, A.F.; Adolphs, N.; et al. De novo mutations in slc25a24 cause a craniosynostosis syndrome with hypertrichosis, progeroid appearance, and mitochondrial dysfunction. Am. J. Hum. Genet. 2017, 101, 833–843.
- Cavero, S.; Traba, J.; Del Arco, A.; Satrústegui, J. The calcium-dependent atp-mg/pi mitochondrial carrier is a target of glucose-induced calcium signalling in saccharomyces cerevisiae. Biochem. J. 2005, 392, 537–544.
- Harborne, S.P.D.; King, M.S.; Crichton, P.G.; Kunji, E.R.S. Calcium regulation of the human mitochondrial atp-mg/pi carrier slc25a24 uses a locking pin mechanism. Sci. Rep. 2017, 7, 45383.
- Traba, J.; Satrústegui, J.; del Arco, A. Characterization of scamc-3-like/slc25a41, a novel calcium-independent mitochondrial atp-mg/pi carrier. Biochem. J. 2009, 418, 125–133.
- Hoffman, N.E.; Chandramoorthy, H.C.; Shanmughapriya, S.; Zhang, X.Q.; Vallem, S.; Doonan, P.J.; Malliankaraman, K.; Guo, S.; Rajan, S.; Elrod, J.W.; et al. Slc25a23 augments mitochondrial Ca2⁺ uptake, interacts with mcu, and induces oxidative stress-mediated cell death. Mol. Biol. Cell 2014, 25, 936–947.
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18.
- Nguyen, T.T.; Quan, X.; Hwang, K.H.; Xu, S.; Das, R.; Choi, S.K.; Wiederkehr, A.; Wollheim, C.B.; Cha, S.K.; Park, K.S. Mitochondrial oxidative stress mediates high-phosphate-induced secretory defects and apoptosis in insulin-secreting cells. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E933–E941.
- Bose, S.; French, S.; Evans, F.J.; Joubert, F.; Balaban, R.S. Metabolic network control of oxidative phosphorylation: Multiple roles of inorganic phosphate. J. Biol. Chem. 2003, 278, 39155–39165.
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ros generation and uncoupling (review). Int. J. Mol. Med. 2019, 44, 3–15.
- Hernansanz-Agustín, P.; Ramos, E.; Navarro, E.; Parada, E.; Sánchez-López, N.; Peláez-Aguado, L.; Cabrera-García, J.D.; Tello, D.; Buendia, I.; Marina, A.; et al. Mitochondrial complex i deactivation is related to superoxide production in acute hypoxia. Redox Biol. 2017, 12, 1040–1051.
- Scialò, F.; Fernández-Ayala, D.J.; Sanz, A. Role of mitochondrial reverse electron transport in ros signaling: Potential roles in health and disease. Front. Physiol. 2017, 8, 428.
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 2016, 167, 457–470.e13.
- Brand, M.D.; Goncalves, R.L.; Orr, A.L.; Vargas, L.; Gerencser, A.A.; Borch Jensen, M.; Wang, Y.T.; Melov, S.; Turk, C.N.; Matzen, J.T.; et al. Suppressors of superoxide-h(2)o(2) production at site i(q) of mitochondrial complex i protect against stem cell hyperplasia and ischemia-reperfusion injury. Cell Metab. 2016, 24, 582–592.
- Selivanov, V.A.; Zeak, J.A.; Roca, J.; Cascante, M.; Trucco, M.; Votyakova, T.V. The role of external and matrix ph in mitochondrial reactive oxygen species generation. J. Biol. Chem. 2008, 283, 29292–29300.


