Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Anna Sacco | + 2973 word(s) | 2973 | 2022-03-22 04:56:10 | | | |
| 2 | Camila Xu | Meta information modification | 2973 | 2022-03-23 04:08:49 | | | | |
| 3 | Camila Xu | Meta information modification | 2973 | 2022-03-24 07:40:08 | | | | |
| 4 | Camila Xu | Meta information modification | 2973 | 2022-03-24 07:40:51 | | | | |
| 5 | Camila Xu | Meta information modification | 2973 | 2022-03-24 07:41:32 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Sacco, A. Regulatory miRNAs and Copper. Encyclopedia. Available online: https://encyclopedia.pub/entry/20877 (accessed on 26 July 2026).
Sacco A. Regulatory miRNAs and Copper. Encyclopedia. Available at: https://encyclopedia.pub/entry/20877. Accessed July 26, 2026.
Sacco, Anna. "Regulatory miRNAs and Copper" Encyclopedia, https://encyclopedia.pub/entry/20877 (accessed July 26, 2026).
Sacco, A. (2022, March 22). Regulatory miRNAs and Copper. In Encyclopedia. https://encyclopedia.pub/entry/20877
Sacco, Anna. "Regulatory miRNAs and Copper." Encyclopedia. Web. 22 March, 2022.
Copy Citation
Non-coding RNAs (ncRNAs), including microRNAs (miRNAs), are key regulators of differentiation and development. In the cell, transcription factors regulate the production of miRNA in response to different external stimuli. Copper (Cu) is a heavy metal and an essential micronutrient with widespread industrial applications. It is involved in a number of vital biological processes encompassing respiration, blood cell line maturation, and immune responses.
Alzheimer’s disease
cardiovascular disease
microRNAs
copper
1. Introduction
Copper (Cu) is an essential transition metal present in traces in our body, and either a Cu deficiency or excess is life-threatening.
A number of studies have indicated that an imbalance in Cu levels and a high burden of oxidative stress are contributors to Alzheimer’s disease (AD), the main form of dementia in the elderly [1][2].
It is known that Cu is a cofactor in the regulation of a number of processes associated with hypoxia. This condition is triggered by an insufficient oxygen supply to tissues and organs that results from pathological conditions underlying AD and ischemic cardiovascular disease (CVD). Hypoxia response is mainly regulated by the hypoxia-inducible factor (HIF) family of oxygen-sensitive transcription factors [3], including HIF-1α, which is regulated by Cu [4].
HIF-1 regulates oxygen homeostasis by modulation of angiogenesis and vascular remodeling, as well as oxygen utilization. HIF also regulates glucose metabolism and redox homeostasis and plays a critical protective role in tissue response to hypoxia of ischemic heart disease, as well as myocardial infarction (MI) [5].
Under the condition of low oxygen levels, HIF-1 is not degraded but acts as a transcriptional target underlying several processes via hypoxia response elements (HREs) at the binding sites, defined as HBSs. Other factors modifying HIF-1′s ability to bind to HREs include epigenetic modifications such as CpG methylation and DNA damage caused by oxidative stress due to elevated intracellular levels of reactive oxygen species (ROS) [6].
The main pathological process triggering the onset of ischemic heart disease is an inflammatory process in which metabolic cells are activated, thereby increasing oxygen demand.
Cu is highly involved in inflammatory processes, either through ceruloplasmin, an acute phase reactant, or through non-ceruloplasmin Cu (also known as “free Cu”), since both increase during inflammation [7]. Despite the accumulated knowledge and technological achievements in amyloid beta (Aβ) and tau detection which build up in the AD brain and typify AD pathology and the new FDA-accelerated approval of the controversial disease-modifying drug Aducanumab (marketed as Aduhelm), which likely can reasonably translate to a clinical benefit [8], the current consensus in the field is that the cause of AD is incompletely defined. An alternative and valid approach toward AD should consider the disease as a multifactorial disorder where many risk factors contribute to global AD susceptibility. In this complex interplay, aging and oxidative stress—mainly through transition metal imbalance—constitute the leading risk factors [1][9]. Oxidative stress is a central molecular mechanism underlying transition metal-induced toxicity and hypoxia. During brain hypoxia, the autophagy machinery may abnormally accumulate high amounts of transition metals such as Cu and iron (Fe), which are normally present in the brain since they are necessary for correct brain functioning. When not bound to proteins or enzymes, Fe and Cu undergo redox cycling reactions with H2O2 (Fenton-type reactions), resulting in the production of ROS, of which H2O2 can diffuse through the cell membrane and then produce the very reactive hydroxyl radical (HO•) catalyzed by Cu and Fe (Fenton-type reactions) [10], eventually leading to tissue damage. The picture of Cu imbalance in AD emerging from a recent meta-analysis [11] is consistent with a shift or displacement of the metal from functional bound Cu to a labile toxic non-ceruloplasmin Cu pool that can easily cross the blood–brain barrier, affecting the aggregation of Aβ and likely producing oxidative stress [1][9].
Consistently, the literature has pointed at a brain-to-heart connection, focusing mainly on the associations between neurodegeneration and cerebral hypoperfusion, hypertension, genetic risk factors (APOE4 and MTHFR gene mutations), high cholesterol, diabetes mellitus, obesity [12], and more recently Cu imbalance [13].
It is known that severe but also prolonged hypoxia is involved in AD neurodegeneration, as one third of stroke patients suffer from post-stroke dementia [14]. Despite this body of literature, the mechanisms linking AD and CVD—which may mediate heart failure (HF), MI, coronary artery disease, atrial fibrillation, and vasculopathy—are still not completely understood.
Among the newly identified mechanisms of Cu toxicity in AD, there is the loss of endothelial lipoprotein receptor-related protein 1 (LRP1), which has been shown to cause aberrant parenchymal Aβ buildup in various AD mouse models [15][16] mainly in a process that appears to be orchestrated by microRNAs [16]. As a matter of the fact, increasing evidence shows that hypoxia resulting from pathological conditions associated with AD and CVD induces the expression of a subset of microRNA (miRNAs) called hypoxia-induced miRNAs [3], which act as regulators of cell responses to a drop in oxygen tension.
miRNAs are short, single-stranded, non-coding RNA molecules of 19–25 nucleotides whose activity is crucial in gene silencing [17]. MiRNAs are an important epigenetic component, as miRNA expression is controlled by epigenetic modifications in a tissue-specific manner at the transcriptional level [18]. In the cytoplasm, miRNAs bind messenger RNAs (mRNAs) in a complementary way. This bond induces gene silencing through the inhibition of translation or degradation of mRNA [19]. One miRNA can influence the expression of hundreds of genes, and each mRNA molecule can be regulated by different miRNAs [20]. Most miRNAs are localized intracellularly but can be released into the blood’s circulation and travel to different districts, thus participating in cell–cell communication processes [21].
miRNAs are also present in the nucleus. Although the function of nuclear miRNAs has not been fully elucidated, the results of several studies show that nuclear miRNAs are involved in gene silencing and gene activation [22]. Most of the functions of miRNAs are linked to recognition by specific regions called miRNA response elements (MREs), which form the interaction link between miRNA and mRNA [23].
MiRNAs, together with ROS and other cell-signaling species such as NF-κB, a family of transcription factors, act as nodes of cross-talk in the inflammation cascade [24]. Several studies have revealed how specific miRNAs are regulated by HIF during hypoxic conditions, thus establishing a link between HIF, miRNA, and CVD states [25]. In the same line of thinking, since HIF is strictly linked to Cu regulation, HIF can be proposed as the possible missing link between Cu and miRNA regulation in AD, which is known to be associated with Cu imbalance [11] and cardiovascular risk.
2. Non-Coding RNAs and Cu Metabolism in Physiology
Non-coding RNAs (ncRNAs) are key regulators of differentiation and development. In 2003, the Human Genome Project (HGP) was completed, and about 20,000–25,000 genes were identified, offering the opportunity to predict the outcomes of individuals diagnosed or threatened with complex diseases such as AD [26].
Cu is a heavy metal and an essential micronutrient with widespread industrial applications. As an essential metal, it is involved in a number of vital biological processes encompassing respiration, blood cell line maturation, immune responses, wound healing, myelin sheath formation, and neurotransmitter synthesis and regulation. It is indispensable for brain and heart development and correct physiology. Severe Cu deficiencies can cause cardiac, bone, immune, and central nervous system conditions, while Cu chronic excess, exposure, or displacements in tissues and organs are associated with liver damage and neurodegenerative disorders [27].
The interplay between miRNA regulation and Cu in physiology will be discussed herein.
2.1. Role of microRNAs in Cell Regulation in Physiology
The genome consists of a large fraction of sequences of RNAs that do not encode any proteins [28] according to Palazzo and collaborators. It is estimated that 99% of the total RNA species present in mammalian cells are non-coding RNAs [29].
The ncRNAs can be classified by length (small: 18–200 nt; long: >200 nt) [30] or by function. The housekeeping ncRNAs include tRNAs and rRNAs, while the regulatory transcripts comprise miRNAs [31]. The miRNAs are a group of small ncRNA molecules.
The biogenesis of miRNAs involves being encoded by genomic DNA with the action of RNA polymerase II, which generates the primary RNA (pri-miRNA). This first product is usually several kilobases long. Then, the pri-miRNA is processed by RNase III Drosha into short hairpin structures called pre-miRNAs, which are exported from the nucleus by exportin. In the cytoplasm, the RNase III enzyme Dicer cuts the pre-miRNAs into mature miRNAs composed of about 22 bases [26]. In association with the Argonaute (AGO) proteins, miRNAs form the RNA-induced silencing complex (RISC), which allows for the translational repression or degradation of the target mRNA. The action of the miRNA on the mRNA depends on the complementarity of the miRNA with its target. When the complementarity is imperfect, the result is translational repression [32], but target RNA destabilization is also observed very often.
According to recent studies, the regulation of miRNA functionality and expression is controlled at three different levels: transcription, processing, and subcellular localization.
In the cell, the transcription factors regulate the production of miRNAs in response to different external stimuli such as inflammation. This process permits control at the transcriptional level [33].
There are several regulatory mechanisms that control miRNA maturation at various stages, starting from the primary transcript. Some of these factors, are the SMAD proteins [34] and Arsenate-resistance protein 2 (ARS2), which permit processing of pri-miRNA [35]. In addition, the tumor suppressor protein p53 has an important role in miRNA processing. The link between p53 and the protein Drosha increases the processing of pri-miRNA to pre-miRNA, and this interaction induces the maturation of specific miRNAs. The levels of p53 in turn can be negatively or positively regulated by specific miRNAs [36][37].
The processing of miRNAs can be post-transcriptionally regulated by RNA-binding proteins (RBPs), which can recognize some regions of miRNA precursors and modulate the processing efficiency. One of these regulators is the stem cell factor LIN28, which interacts with the pre-miRNAs and blocks their expression [38]. Another regulatory factor is HNRNPA1, which binds to pri-miR-18a and increases the function of Drosha [39]. Nussbacher and collaborators found that 92% of RBPs interact directly with one miRNA locus, and they are cell line specific [40].
miRNA is the most studied class of ncRNA. miRNAs recognize their targets through miRNA response elements (MREs). Generally, the most conserved binding sites are enriched in the 3′UTR sequences, but MREs can also be present in the 5′ UTR sequences as well as in the coding regions of mRNAs [41]. miRNAs form a complex (RISC) by binding to various AGO proteins, allowing the miRNAs to recognize their targets and to bind and detach easily from targets. Furthermore, AGO proteins are important cofactors for the binding of RISC with the target, and different types of AGO proteins mediate different gene regulation [42].
2.2. Cu Intake in Human Diet and Drinking Water
Cu balance is established by the rates of dietary absorption from food, supplements, drinking water, and excretion through stools and bile, and it is tightly controlled (Figure 1).
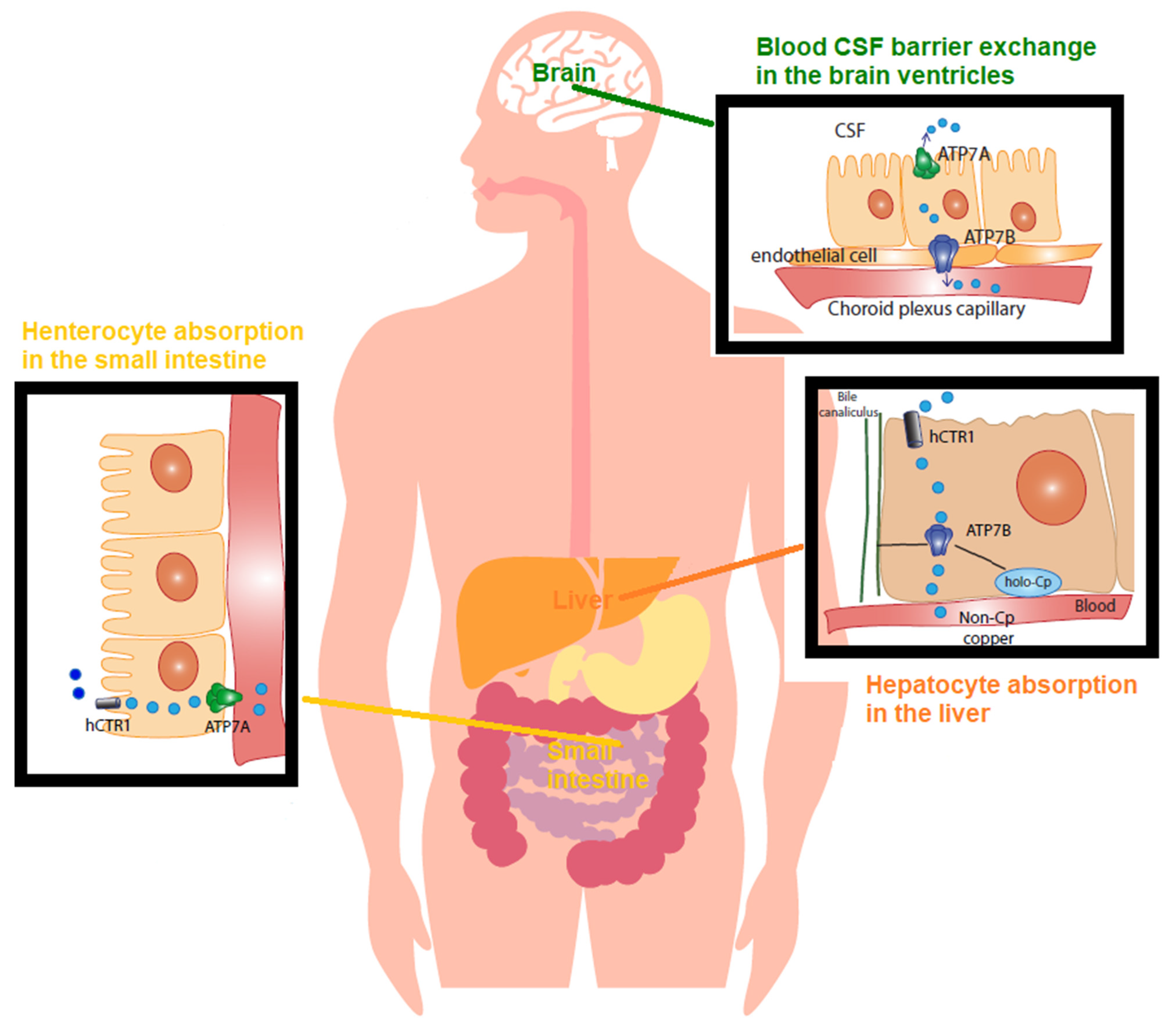
Figure 1. Copper (Cu) in physiology The recommended dietary allowance (RDA) for Cu is 0.9–1.3 mg/day. This represents the intake level sufficient to meet the nutrient requirements. Accordingly, humans normally ingest 1.5 mg/day of Cu via beverage and foods. The Cu balance is determined by the equilibrium between the rates of dietary absorption from diet and excretion through stools and bile, and it is tightly regulated. In the small bowel, concerning Cu absorption into the enterocyte, Cu2+ is reduced by reductases. CTR1 imports Cu1+ within the cell. Cu is absorbed as a pool of low molecular weight soluble complexes and pumped out of the enterocyte’s basolateral membrane by the Cu-transporting P-type ATPase (ATPase7A) via the vesicular compartment (not shown). Cu is then transported, mostly bound to amino acids, peptides, micronutrients, and albumin, and transported into the serum. From the gut, this pool of low molecular weight Cu, known as non-ceruloplasmin (non-Cp) Cu, travels to the liver through the portal vein. The liver represents the main organ of storage and utilization of Cu. CTR1 facilitates Cu intake in the hepatocytes and delivery to chaperones. In the liver, ATPase7B, the homologue of enterocytes’ ATPase7A, incorporates Cu into ceruloplasmin. Of the Cu, 75–95% tightly binds to ceruloplasmin, whereas the remainder loosely binds to and is exchanged among albumin, α2 macroglobulin, amino acids, peptides, and several micronutrients. Hepatocytes limit the non-Cp Cu concentration in the blood to 0.008–1.6 µmol/L (equivalent to 0.05–1 mg/dL), which is the upper limit of the normal reference range of non-Cp Cu in serum after an overnight fast. An excess of Cu induces the translocation of ATPase7B from the trans-Golgi network to the canalicular membrane (via the vesicular compartment), where the metal is released into the bile. As for blood barrier exchange in the brain ventricles, the endothelial cells of the brain’s capillaries constitute the blood–brain barrier (BBB). The cerebrospinal fluid (CSF) is the biological fluid that surrounds the brain and fills the brain ventricles, and it is secreted by the choroid plexus. The Cu in CSF has values in the range of 0.5–2.5 µmol/L. In the choroid plexus, non-Cp Cu is the main form of Cu taken up from the blood and is then released into the brain by processes mediated by CTR1, ATPase7A, and ATPase7AB.
The Cu recommended dietary allowance (RDA)—the intake level sufficient to meet the nutrient requirements for 97–98% of people—is 0.9–1.3 mg/day (United States (US)) [47]. On average, the individual consumes ~2 mg per day (WHO, 1996) [48], and 2–3 mg/day of Cu intake is safe and adequately prevents Cu deficiency, while ingesting >5 mg/day is deemed toxic [49].
2.3. Cu Regulation in Physiology
The Cu from a diet is readily absorbed from the stomach and intestines (duodenum and ileum) at a rate of 0.5 mg/day [50]. Then, it is transported to the liver (Figure 1), and 0.5 mg/day generally corresponds to 30% of the intake (75% from food and 25% from supplements and beverages). The remaining amount of Cu is directly eliminated through the “mucosal block”, consisting of metallothioneins (MTs), which are cysteine-rich proteins that bind and sequester metal ions and are located in the enterocyte cells lining the mucosal tract, which trap Cu and facilitate its excretion [50]. The rate of Cu absorption lessens as the Cu intake rises. The dietary Cu intake changes from 56% during a low-Cu regimen to 12% under a high-Cu diet [51]. Even though in a high-Cu intake diet the efficiency of Cu absorption declines, more Cu is absorbed and retained [51].
In the stomach, duodenum, and ileum, Cu is absorbed as a pool of low molecular-weight-soluble complexes of Cu [52]. In the intestinal lining cells, Cu is then pumped out of the enterocyte by the Cu-transporting P-type ATPase (Cu-ATPase) 7A (ATPase7A) located at the basolateral membrane. This pool of Cu is called non-ceruloplasmin Cu, a serum Cu2+ species not structurally bound to proteins and primarily not bound to ceruloplasmin, the main Cu protein in general circulation [53]. Non-ceruloplasmin Cu is mostly loosely bound to low molecular weight compounds such as amino acids, peptides, and micronutrients, and it is exchanged among them, albumin, and α2-macroglobulin and transported into the serum. From the gut, it reaches the liver through the portal vein. In physiology, hepatocytes take up most of this Cu species and regulate the blood’s non-ceruloplasmin Cu concentration in the range of 0.008–1.6 µmol/L (equivalent to 0.05–1 mg/dL). These are the normal reference range values of non-ceruloplasmin Cu in serum after an overnight fast [50][54] (Figure 1).
In the liver, namely in the interstitial fluid surrounding cells, human Cu transporter 1 (CTR1) regulates Cu(I) entry into the hepatocyte, and Cu chaperons and transporters then accompany the metal to the sites of its utilization within the cell [55]. At the trans-Golgi network, Cu is packed into Cu proteins and enzymes. The main pathways include cytochromes in the mitochondria for oxidative respiration (cytochrome oxidase (COX)), copper/zinc superoxide dismutase (Cu/Zn SOD) for antioxidant defenses, and ceruloplasmin for controlling the iron (Fe) oxidative state [55].
Similar to ATPase7A, the Cu-transporting P-type ATPase (Cu-ATPase) 7B (ATPase7B) is a key protein controlling the Cu balance. It is a Cu pump located in the trans-Golgi network that loads Cu into nascent ceruloplasmin in the hepatocyte [55] and facilitates Cu excretion into the bile when the metal exceeds the needs of the cell [50] (Figure 1). Ceruloplasmin binds 75–85% of circulating Cu, whereas the remainder constitutes the non-ceruloplasmin Cu species [55].
In correct physiology, Cu in the CSF, the biological fluid that surrounds the brain and is produced and secreted by the choroid plexus, ranges between 0.5 and 2.5 µmol/L [56]. Perfusion of three species of radioactive 64Cu (a homolog of non-ceruloplasmin 64Cu, 64Cu-albumin, and 64Cu-ceruloplasmin) into a rat brain via the internal carotid artery demonstrated that non-ceruloplasmin 64Cu in a rat choroid plexus is the main species taken up into the brain, being about 50 times higher than Cu-albumin and 1000 times higher than Cu-ceruloplasmin [57]. This is also exemplified in Wilson’s disease, the paradigmatic disorder for non-ceruloplasmin Cu toxicosis or accumulation, in which non-ceruloplasmin Cu easily crosses the blood–brain barrier (BBB) [50], and it has also been found in AD [58].
Wilson’s disease is a rare genetic disorder caused by mutations in the ATP7B gene, encoding for the ATPase7B the Cu pump that handles Cu incorporation into ceruloplasmin and Cu excretion through the bile. ATP7B mutations cause Cu accumulation in the liver, kidneys, and other parenchymal tissues, including the brain [59]. Cu deposition in the cornea and around the iris of the eye, known as Kayser–Fleischer rings (for a specialized review, refer to [60]), is typical. Wilson’s disease is also typified by levels of non-ceruloplasmin Cu higher than the 1.6 µmol/L that is considered toxic [61]. Aside from eating food and supplements and drinking water, Cu exposure can also occur through breathing air or by skin contact with soil, water, and other Cu-containing substances, including nanoparticles [62].
References
- Squitti, R.; Faller, P.; Hureau, C.; Granzotto, A.; White, A.R.; Kepp, K.P. Copper Imbalance in Alzheimer’s Disease and Its Link with the Amyloid Hypothesis: Towards a Combined Clinical, Chemical, and Genetic Etiology. J. Alzheimer’s Dis. 2021, 83, 23–41.
- Squitti, R.; Siotto, M.; Polimanti, R. Low-copper diet as a preventive strategy for Alzheimer’s disease. Neurobiol. Aging 2014, 35, S40–S50.
- Ziello, J.E.; Jovin, I.S.; Huang, Y. Hypoxia-Inducible Factor (HIF)-1 Regulatory Pathway and its Potential for Therapeutic Intervention in Malignancy and Ischemia. Yale J. Biol. Med. 2007, 80, 51–60.
- Feng, W.; Ye, F.; Xue, W.; Zhou, Z.; Kang, Y.J. Copper Regulation of Hypoxia-Inducible Factor-1 Activity. Mol. Pharmacol. 2008, 75, 174–182.
- Semenza, G.L. Hypoxia-Inducible Factor 1 and Cardiovascular Disease. Annu. Rev. Physiol. 2014, 76, 39–56.
- Rodriguez, H.; Drouin, R.; Holmquist, G.P.; Akman, S.A. A Hot Spot for Hydrogen Peroxide-Induced Damage in the Human Hypoxia-Inducible Factor 1 Binding Site of thePGK 1Gene. Arch. Biochem. Biophys. 1997, 338, 207–212.
- Squitti, R.; Siotto, M.; Salustri, C.; Polimanti, R. Metal dysfunction in Alzheimer’s disease. In Studies on Alzheimer’s Disease; Springer: Berlin/Heidelberg, Germany, 2013; pp. 73–97.
- Alexander, G.C.; Knopman, D.S.; Emerson, S.S.; Ovbiagele, B.; Kryscio, R.J.; Perlmutter, J.S.; Kesselheim, A.S. Revisiting FDA Approval of Aducanumab. N. Engl. J. Med. 2021, 385, 769–771.
- Ayton, S.; Portbury, S.; Kalinowski, P.; Agarwal, P.; Diouf, I.; Schneider, J.A.; Morris, M.C.; Bush, A.I. Regional brain iron associated with deterioration in Alzheimer’s disease: A large cohort study and theoretical significance. Alzheimer’s Dement. 2021, 17, 1244–1256.
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464.
- Squitti, R.; Ventriglia, M.; Simonelli, I.; Bonvicini, C.; Costa, A.; Perini, G.; Binetti, G.; Benussi, L.; Ghidoni, R.; Koch, G.; et al. Copper Imbalance in Alzheimer’s Disease: Meta-Analysis of Serum, Plasma, and Brain Specimens, and Replication Study Evaluating ATP7B Gene Variants. Biomolecules 2021, 11, 960.
- Tublin, J.M.; Adelstein, J.M.; Del Monte, F.; Combs, C.K.; Wold, L.E. Getting to the Heart of Alzheimer Disease. Circ. Res. 2019, 124, 142–149.
- Squitti, R.; Mendez, A.; Ricordi, C.; Siotto, M.; Goldberg, R. Copper in Glucose Intolerance, Cognitive Decline, and Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 2019, 33, 77–85.
- Mijajlović, M.D.; Pavlović, A.; Brainin, M.; Heiss, W.-D.; Quinn, T.J.; Ihle-Hansen, H.B.; Hermann, D.M.; Ben Assayag, E.; Richard, E.; Thiel, A.; et al. Post-stroke dementia–a comprehensive review. BMC Med. 2017, 15, 1–12.
- Singh, I.; Sagare, A.P.; Coma, M.; Perlmutter, D.; Gelein, R.; Bell, R.D.; Deane, R.J.; Zhong, E.; Parisi, M.; Ciszewski, J.; et al. Low levels of copper disrupt brain amyloid-β homeostasis by altering its production and clearance. Proc. Natl. Acad. Sci. USA 2013, 110, 14771–14776.
- Hsu, H.-W.; Rodriguez-Ortiz, C.J.; Lim, S.L.; Zumkehr, J.; Kilian, J.G.; Vidal, J.; Kitazawa, M. Copper-Induced Upregulation of MicroRNAs Directs the Suppression of Endothelial LRP1 in Alzheimer’s Disease Model. Toxicol. Sci. 2019, 170, 144–156.
- Liu, B.; Li, J.; Cairns, M.J. Identifying miRNAs, targets and functions. Brief. Bioinform. 2012, 15, 1–19.
- Abdelfattah, A.M.; Park, C.; Choi, M.Y. Update on non-canonical microRNAs. Biomol. Concepts 2014, 5, 275–287.
- Wahid, F.; Shehzad, A.; Khan, T.; Kim, Y.Y. MicroRNAs: Synthesis, mechanism, function, and recent clinical trials. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2010, 1803, 1231–1243.
- Bronze-Da-Rocha, E. MicroRNAs Expression Profiles in Cardiovascular Diseases. BioMed Res. Int. 2014, 2014, 985408.
- Creemers, E.E.; Tijsen, A.J.; Pinto, Y.M. Circulating microRNAs: Novel biomarkers and extracellular communicators in cardiovascular disease? Circ. Res. 2012, 110, 483–495.
- Miao, L.; Yao, H.; Li, C.; Pu, M.; Yao, X.; Yang, H.; Qi, X.; Ren, J.; Wang, Y. A dual inhibition: MicroRNA-552 suppresses both transcription and translation of cytochrome P450 2E1. Biochim. Et Biophys. Acta 2016, 1859, 650–662.
- Nair, A.A.; Tang, X.; Thompson, K.J.; Vedell, P.T.; Kalari, K.R.; Subramanian, S. Frequency of MicroRNA Response Elements Identifies Pathologically Relevant Signaling Pathways in Triple-Negative Breast Cancer. iScience 2020, 23, 101249.
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86.
- Serocki, M.; Bartoszewska, S.; Janaszak-Jasiecka, A.; Ochocka, R.J.; Collawn, J.F.; Bartoszewski, R. miRNAs regulate the HIF switch during hypoxia: A novel therapeutic target. Angiogenesis 2018, 21, 183–202.
- Li, M.; Marin-Muller, C.; Bharadwaj, U.; Chow, K.-H.; Yao, Q.; Chen, C. MicroRNAs: Control and Loss of Control in Human Physiology and Disease. World J. Surg. 2008, 33, 667–684.
- Kepp, K.P.; Squitti, R. Copper imbalance in Alzheimer’s disease: Convergence of the chemistry and the clinic. Co-Ord. Chem. Rev. 2019, 397, 168–187.
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15 (Suppl. 1), R17–R29.
- Palazzo, A.F.; Lee, E.S. Non-coding RNA: What is functional and what is junk? Front. Genet. 2015, 6, 2.
- Dhanoa, J.K.; Sethi, R.S.; Verma, R.; Arora, J.S.; Mukhopadhyay, C.S. Long non-coding RNA: Its evolutionary relics and biological implications in mammals: A review. J. Anim. Sci. Technol. 2018, 60, 25.
- Romano, G.; Veneziano, D.; Acunzo, M.; Croce, C.M. Small non-coding RNA and cancer. Carcinogenesis 2017, 38, 485–491.
- O’Connell, R.M.; Rao, D.S.; Chaudhuri, A.A.; Baltimore, D. Physiological and pathological roles for microRNAs in the immune system. Nat. Rev. Immunol. 2010, 10, 111–122.
- Chatterjee, S.; Grosshans, H. Active turnover modulates mature microRNA activity in Caenorhabditis elegans. Nature 2009, 461, 546–549.
- Davis-Dusenbery, B.; Hilyard, A.C.; Lagna, G.; Hata, A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature 2008, 454, 56–61.
- Gruber, J.J.; Zatechka, D.S.; Sabin, L.R.; Yong, J.; Lum, J.J.; Kong, M.; Zong, W.-X.; Zhang, Z.; Lau, C.-K.; Rawlings, J.; et al. Ars2 Links the Nuclear Cap-Binding Complex to RNA Interference and Cell Proliferation. Cell 2009, 138, 328–339.
- Abdi, J.; Rastgoo, N.; Li, L.; Chen, W.; Chang, H.; Abdi, J.; Rastgoo, N.; Li, L.; Chen, W.; Chang, H. Role of tumor suppressor p53 and micro-RNA interplay in multiple myeloma pathogenesis. J. Hematol. Oncol. 2017, 10, 169.
- Feng, Z.; Zhang, C.; Wu, R.; Hu, W. Tumor suppressor p53 meets microRNAs. J. Mol. Cell Biol. 2011, 3, 44–50.
- Treiber, T.; Treiber, N.; Plessmann, U.; Harlander, S.; Daiß, J.-L.; Eichner, N.; Lehmann, G.; Schall, K.; Urlaub, H.; Meister, G. A Compendium of RNA-Binding Proteins that Regulate MicroRNA Biogenesis. Mol. Cell 2017, 66, 270–284.e13.
- Michlewski, G.; Guil, S.; Cáceres, J.F. Stimulation of pri-miR-18a processing by hnRNP A1. Regul. Micrornas 2010, 700, 28–35.
- Nussbacher, J.K.; Yeo, G.W. Systematic Discovery of RNA Binding Proteins that Regulate MicroRNA Levels. Mol. Cell 2018, 69, 1005–1016.e7.
- Lee, I.; Ajay, S.S.; Yook, J.I. New class of microRNA targets containing simultaneous 5′-UTR and 3′-UTR interaction sites. Genome Res. 2009, 19, 1175–1183.
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in Control of Gene Expression: An Overview of Nuclear Functions. Int. J. Mol. Sci. 2016, 17, 1712.
- Meijer, H.A.; Kong, Y.W.; Lu, W.T.; Wilczynska, A.; Spriggs, R.V.; Robinson, S.W.; Godfrey, J.D.; Willis, A.E.; Bushell, M. Translational Repression and eIF4A2 Activity Are Critical for MicroRNA-Mediated Gene Regulation. Science 2013, 340, 82–85.
- Gagnon, K.T.; Li, L.; Chu, Y.; Janowski, B.A.; Corey, D.R. RNAi Factors Are Present and Active in Human Cell Nuclei. Cell Rep. 2014, 6, 211–221.
- Wu, H.; Sun, S.; Tu, K. A splicing-independent function of SF2/ASF in microRNA processing. Mol. Cell 2010, 38, 67–77.
- Gebert, L.F.R.; Macrae, I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37.
- DeVita, M.A.; Aulisio, M.P. The Ethics of Medical Mistakes: Historical, Legal, and Institutional Perspectives. Kennedy Inst. Ethic-J. 2001, 11, 115–116.
- World Health Organization. Trace Elements in Human Nutrition and Health; World Health Organization: Geneva, Switzerland, 1996.
- de Romaña, D.L.; Olivares, M.; Uauy, R.; Araya, M. Risks and benefits of copper in light of new insights of copper homeostasis. J. Trace Elem. Med. Biol. 2011, 25, 3–13.
- Bertrand, E.; Lewandowska, E.; Szpak, G.M.; Hoogenraad, T.; Blaauwgers, H.G.; Członkowska, A.; Dymecki, J. Neuropathological analysis of pathological forms of astroglia in Wilson′s disease. Folia Neuropathol. 2001, 39, 73–79.
- Turnlund, J.R.; Keyes, W.R.; Kim, S.K.; Domek, J.M. Long-term high copper intake: Effects on copper absorption, retention, and homeostasis in men. Am. J. Clin. Nutr. 2005, 81, 822–828.
- Kim, H.-J.; Lee, J.Y.; Paik, K.-W.; Koh, K.-W.; Won, J.; Choe, S.; Lee, J.; Moon, J.-T.; Park, Y.-J. Effects of Cu/Al intermetallic compound (IMC) on copper wire and aluminum pad bondability. IEEE Trans. Compon. Packag. Technol. 2003, 26, 367–374.
- Siotto, M.; Squitti, R. Copper imbalance in Alzheimer’s disease: Overview of the exchangeable copper component in plasma and the intriguing role albumin plays. Co-Ord. Chem. Rev. 2018, 371, 86–95.
- Walshe, J.M. Wilson′s disease: The importance of measuring serum caeruloplasmin non-immunologically. Ann. Clin. Biochem. Int. J. Lab. Med. 2003, 40, 115–121.
- Sensi, S.L.; Granzotto, A.; Siotto, M.; Squitti, R. Copper and Zinc Dysregulation in Alzheimer’s Disease. Trends Pharmacol. Sci. 2018, 39, 1049–1063.
- Bucossi, S.; Ventriglia, M.; Panetta, V. Copper in Alzheimer’s disease: A meta-analysis of serum, plasma, and cerebrospinal fluid studies. J. Alzheimer’s Dis. 2011, 24, 175–185.
- Choi, B.-S.; Zheng, W. Copper transport to the brain by the blood-brain barrier and blood-CSF barrier. Brain Res. 2009, 1248, 14–21.
- Squitti, R.; Barbati, G.; Rossi, L.; Ventriglia, M.; Forno, G.D.; Cesaretti, S.; Moffa, F.; Caridi, I.; Cassetta, E.; Pasqualetti, P.; et al. Excess of nonceruloplasmin serum copper in AD correlates with MMSE, CSF-amyloid, and h-tau. Neurology 2006, 67, 76–82.
- Zischka, H.; Einer, C. Mitochondrial copper homeostasis and its derailment in Wilson disease. Int. J. Biochem. Cell Biol. 2018, 102, 71–75.
- Bandmann, O.; Weiss, K.H.; Kaler, S.G. Wilson′s disease and other neurological copper disorders. Lancet Neurol. 2015, 14, 103–113.
- Roberts, E.A.; Schilsky, M.L.; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: An update. Hepatology 2008, 47, 2089–2111.
- Ameh, T.; Sayes, C.M. The potential exposure and hazards of copper nanoparticles: A review. Environ. Toxicol. Pharmacol. 2019, 71, 103220.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
849
Revisions:
5 times
(View History)
Update Date:
24 Mar 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No