 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | antje anji | + 4486 word(s) | 4486 | 2022-03-17 06:46:00 | | | |
| 2 | Lindsay Dong | -2 word(s) | 4484 | 2022-03-21 02:59:00 | | |
Video Upload Options
Exosomes of endosomal origin are one class of extracellular vesicles that are important in intercellular communication. Exosomes are released by all cells inbody and their cargo consisting of lipids, proteins and nucleic acids has a footprint reflective of their parental origin. The exosomal cargo has the power to modulate the physiology of recipient cells in the vicinity of the releasing cells or cells at a distance. Harnessing the potential of exosomes relies upon the purity of exosome preparation. Exosomes have an intercellular communicator role in the spread of misfolded proteins aiding the propagation of pathology.
1. Introduction
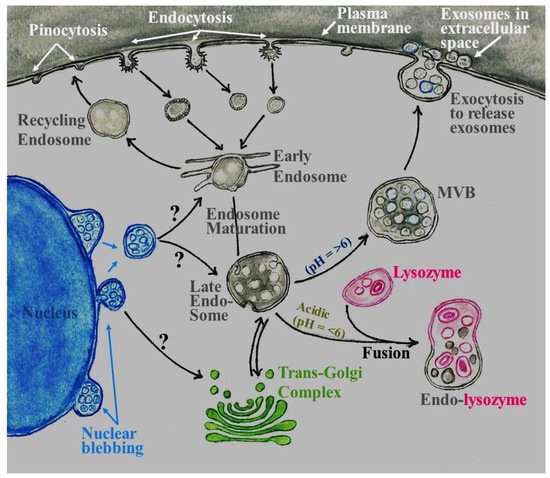
2. Role of Exosomes in the Central Nervous System (CNS)
3. Role of Exosomes in Neurodegenerative Diseases
Alzheimer’s Disease: Alzheimer’s disease is the most common form of neurodegenerative dementia characterized by a progressive loss of memory and cognitive abilities. Central to the pathology of Alzheimer’s disease is formation of extracellular aggregates of β-amyloid (Aβ) known as amyloid plaques combined with neurofibrillary tangles of tau. The (Aβ) peptides are derived from sequential proteolytic processing of amyloid precursor protein (APP) by β- and γ-secretases. As APP is an intracellular protein, a hypothesis was formulated that the pathological lesions of neurodegenerative disease involves the physical spread of the misfolded protein from neuron to neuron [45]. However, mechanism(s) for transmission of misfolded proteins remained an intriguing question. One of the earliest reports that started to shed light on the possible mechanisms how Aβ is shed into the extracellular space came from the study conducted by Rajendran and Colleagues [46]. While investigating the location of APP cleavage, they observed that β-secretase cleavage of APP occurs in a subset of early endosomes with subsequent trafficking of Aβ peptide to multivesicular bodies. A small fraction of Aβ peptide associated with exosome membrane was secreted into the extracellular space. Exosomes containing amyloid plaques had exosomal marker proteins, flotillin-1 and Alix. Thus, exosome membrane-associated Aβ peptide may represent a novel mechanism that contributes to amyloid plaque formation in the extracellular space [46]. Since this initial observation, full-length APP and many of its metabolites and several members of secretase family of proteases involved in APP processing were detected in exosomes [47]. Aβ can exist in different conformational states that have different properties and intermediate products of fibril formation. Of these, low-molecular-weight Aβ and protofibrils have been suggested to be particularly neurotoxic and act as seeds for protein aggregation [48][49]. Exosomes isolated from postmortem brains of Alzheimer’s disease patients were shown to have increased levels of Aβ oligomers. These exosomes were internalized when incubated with cultured neurons. Most importantly, they were able to spread Aβ oligomers to other neurons, causing cytotoxicity [50]. The initial observations were confirmed by incubating exosomes containing APP with primary cultures of normal neurons in vitro [51] and in vivo [52]. The question is why are Aβ or Aβ oligomers packaged into exosomes? Do neurons sense the toxic nature of Aβ or Aβ oligomers and hence try to clear toxic proteins from intracellularly present Aβ or Aβ oligomers similar to transferrin receptor in reticulocytes [53][54]? Monoubiquitination is required for sorting into MVB/exosomes. That raises a second question as to whether Aβ undergoes ubiquitination to be sorted into MVB/exosomes. The amyloid precursor protein (APP) has five lysine residues (Lys-724, Lys-725, Lys-726, Lys-751, and Lys-763) at its C-terminal end [55]. These residues have been mutated individually or in combination to examine effect on APP processing to form Aβ peptide. Ubiquitination of APP at Lys-726 mediated by the F-box and leucine-rich repeat protein2 (FBL2), a component of the E3 ubiquitin ligase, reduced Aβ generation [56]. On the other hand, APP ubiquitination at Lys-763 sequestered APP in the Golgi complex and prevented APP maturation [57]. Inhibition of ubiquitination by substitution of all five lysine residues to arginine in C-terminal fragment of APP (C99) prevented efficient degradation of APP and accumulation of protein in structures with Golgi-like appearance. This was attributed to a deficiency in endoplasmic reticulum-associated degradation. The C99 undergoes cleavage by γ-secretase to produce Aβ [58]. Mutation of three lysine residues (Lys-724, Lys-725, and Lys-726) simultaneously caused the protein to be retained in the limiting membrane of endosomes instead of becoming internalized into intraluminal vesicles of MVBs [59]. When all five lysine residues were mutated to prevent ubiquitination, the protein did not efficiently sort to MVB/exosomes and a selective increase in Aβ40 was observed [60]. This finding is comparable to the presence of Aβ40 in amyloid deposits in cerebral amyloid angiopathy [61]. While it is evident that ubiquitination of APP may direct the protein to MVB/exosomes, direct evidence using neuronal cultures or in vivo models is required to prove that APP, Aβ, or Aβ oligomers are monoubiquitinated for their targeting to MVB/exosomes and that they are not ubiquitinated to be targeted for endoplasmic reticulum-associated degradation (ERAD).
Another pathological hallmark of Alzheimer’s disease is abnormally phosphorylated tau protein in neurofibrillary tangles (NFT). Tau is a cytoplasmic protein known to stabilize microtubules. Increasing evidence suggests that the pathological tau protein can spread between cells, recruiting native tau to form aggregates. Microglia phagocytose tau-containing cytopathic neurons and recycle tau through exosomes thus incriminating exosomes in propagation of Alzheimer’s disease. In healthy brains, a number of protein kinases and phosphatases are responsible for phosphorylation and de-phosphorylation of tau, respectively. Dysregulation of these important enzymes may lead to abnormal phosphorylation pattern of tau in AD.
Exosomes released by neurons, astrocytes, and microglia act as scavengers and soak up seed-free soluble Aβ to promote Aβ aggregation that is internalized by microglia for degradation [62][63][64].
Parkinson’s Disease: Parkinson’s disease is one of the most common age-related brain disorders—it is primarily considered a movement disorder, with typical symptoms of a resting tremor, rigidity, bradykinesia and motor instability [65]. Additionally associated with this disease are cognitive decline, depression, and psychosis [66]. Pathologically, it is characterized by degeneration of nigrostriatal dopaminergic neurons and the presence of Lewy bodies which contain misfolded α-synuclein protein in surviving neurons. Alpha synuclein is detected in many body fluids including cerebrospinal fluid and plasma [67][68]. Alpha synuclein is found in culture medium when cells expressing α-synuclein are cultured in vitro [69].
A first indication that exosomes could indeed be involved in the pathogenesis came from an in vitro study employing SH-SY5Y cells expressing α-synuclein. The reserch demonstrated extracellular secretion of α-synuclein via exosomes in a calcium-dependent manner and suggested their involvement in spread of Parkinson’s disease pathology [70]. Following this study, α-synuclein-containing exosomes have been identified from different cells, cerebrospinal fluid, and plasma of Parkinson’s disease patients [71][72][73].
A fundamental question to understand the pathogenesis of Parkinson’s disease is how do exosomes relay the toxic effects of α-synuclein? Exosomes may aid Parkinson’s disease pathogenesis by promoting aggregation of α-synuclein due to their lipid and/or protein composition thus facilitating uptake of α-synuclein by cells. Several studies pointed out that exosomes contain α-synuclein as oligomers. Several cellular processes and enrichment of certain molecules within cells cause α-synuclein oligomerization and often in combination with other proteins. Exosomal ganglioside lipids GM1 or GM3 accelerate α-synuclein aggregation [74]. A combination of ceramides and neurodegeneration-linked proteins including α-synuclein and tau in exosomes is capable of inducing aggregation of wild-type α-synuclein [75]. Oxidation of two adjacent amino acids, methionine [Met(38)] and tyrosine [Tyr(39)], results in aggregation of γ-synuclein and seed aggregation of α-synuclein. Neuronal exosomes containing γ-synuclein upon internalization can cause aggregation of intracellular proteins in astrocytes, resulting in synucleinopathies [76]. Levels of Golgi complex localized the gamma adaptin ear-containing, ARF-binding protein 3 (GGA3) were downregulated in postmortem substantia nigra of PD patients as compared to controls. GGA3 induces oligomerization of α-synuclein in endosomes, resulting in secretion of α-synuclein oligomers [77]. In another study, researchers reported interaction between α-synuclein and the autophagy protein, LC3B that resulted in formation of detergent-insoluble oligomeric aggregates. Alpha-synuclein oligomers are deposited on the surface of late endosomes and are eventually secreted out of human pluripotent stem cells through exosomes [78].
Microglia are a double-edge sword in the CNS as they can be either neuroprotective or neurotoxic. Incubation of microglial cell line BV2 with α-synuclein released an increased number of exosomes enriched with MHC class II molecules and membrane TNFα. Internalization of these exosomes by neurons was neurotoxic suggesting a role for microglia in α-synuclein-induced neurodegeneration [79]. The question is how α-synuclein is internalized by microglia. Microglial cells selectively express Toll-like receptor 2 (TLR2) that acts as a ligand of α-synuclein. Binding of α-synuclein to TLR2 activates microglia. Since α-synuclein is present on the surface of exosomes, they are internalized by microglia. The excessive exosome uptake by microglia causes an inflammatory response [80], inhibition of autophagy, and reduced scavenger activity. Reduced phagocytosis of α-synuclein-containing exosomes was seen in mouse microglia and human monocytes from aged donors. This observation suggests an age-dependent predisposition to incidence of misfolded proteins in Parkinson’s disease [81], which is in line with the onset of Parkinson’s disease occurring predominantly after 60 years of age.
In summary, there has been substantial interest in exosome research in the context of Parkinson’s disease. It is apparent that exosomes are important mediators of α-synuclein transmission among brain cells. In addition, the ability of exosomes to transfer proteins and miRNAs contributes to pathogenesis.
Amyotrophic Lateral Sclerosis: Amyotrophic lateral sclerosis (ALS) is a late-onset, fatal neurodegenerative disease with a median survival of only 2–5 years—it affects upper motor neurons which project from the cortex to brain stem and spinal cord, as well as lower motor neurons that project from the spinal cord to muscles. Patients develop progressive muscle paralysis and death usually occurs due to respiratory failure. Most cases are sporadic but some are familial cases. ALS is characterized by misfolding of Cu/Zn dismutase (SOD-1) [82] and TAR DNA-binding protein 43 (TDP-43) [83]. SOD 1 is a cytosolic mitochondrial enzyme involved in clearance of superoxide molecule, while TDP-43 is a highly conserved nuclear RNA/DNA-binding protein involved in RNA processing. Post-translational modifications such as cleavage, hyper-phosphorylation and ubiquitination of TDP-43 can lead to cytoplasmic accumulation and aggregation of TDP-43. Both SOD-1 and TDP-43 are packaged in exosomes [84][85]. By overexpressing both wild-type and mutated SOD-1 in NSC-34 motor neuron-like cells, Grad and colleagues observed that misfolded SOD-1 protein was transferred from cell to cell via exosomes in addition to direct uptake of SOD-1 protein aggregates by micropinocytosis [85]. Studies have suggested that astrocytes may play a role in pathogenesis of ALS. Exosomes released by primary astrocyte cultures expressing mutant SOD-1 efficiently transferred mutant SOD-1 protein to spinal neurons, causing selective motor neuron death [86]. A study utilizing a SOD-1 transgenic mouse model demonstrated that mutant SOD-1 was enriched in exosomes derived from both neurons and astrocytes, suggesting that these two cell types may contribute to spread of pathology in ALS [87]. TDP-43, another protein involved in pathogenesis of ALS, was detected in exosomes purified from cerebrospinal fluid of ALS patients [88], supporting the idea that exosomes contribute to disease propagation. Indeed, cerebrospinal fluid enriched with TDP-43-containing exosomes was able to promote accumulation of toxic TDP-43 in human glioma U251 cells [89]. Furthermore, TDP-43 oligomers present in exosomes were transmitted intercellularly [90]. Interestingly, levels of exosomal TDP-43 (full-length protein and C-terminal fragments) are upregulated in brains of ALS patients. When Neuro2a cells were exposed to exosomes from ALS brains, TDP-43 was redistributed in the cytoplasm of Neuro2a cells [91]. Compared to other neurodegenerative diseases, research into the pathogenesis of this devastating fatal disease is much more limited. Much of the research has been performed in vitro. With refinement of exosome isolation techniques from brain tissue it is hoped that scholars will have a clearer picture of the role-played by exosomes in spread of ALS.
Exosomes and the blood–brain barrier: The blood–brain barrier is a physical barrier between brain and the peripheral circulation, controlling a strict influx and efflux of molecules to maintain the homeostasis. Accumulating evidence suggests that exosomes have the remarkable ability to cross the blood–brain barrier from both directions. Exosomes carry cargos of membrane and cytosolic proteins and genetic material such as mRNAs, non-coding RNAs including miRNAs that otherwise generally do not cross plasma membrane. Exosomes released from cancer cells have been shown to destroy the blood–brain barrier through action of microRNA-181c, leading to actin mislocalization and perhaps, resulting in breakdown of blood–brain barrier integrity. Such leakiness of the blood–brain barrier is also seen in cases of neurodegeneration often as a result of neuroinflammation. Furthermore, glioblastoma-specific mRNAs have been detected in exosomes in the peripheral circulation [92]. Experimental evidence suggests that exosomes can cross the blood–brain barrier from the periphery and localize in the brain. Analyses of fluorescent or luciferase-labeled exosomes demonstrated that they have the ability to accumulate in the brain from the periphery [93][94]. Exosomes loaded with siRNA were able to deliver their cargo to neurons, microglia and oligodendrocytes in brain when administered intravenously [95]. Exosomes derived from hematopoietic cells can be transferred to Purkinje cells in the brain and importantly were able to modulate gene expression in these cells. This observation suggests that transfer of exosomes via the blood–brain barrier can have functional implications. The ability of exosomes to cross the blood–brain barrier presents a great potential for exosomes as a drug delivery system. Equally important is that uptake of the exosomal cargo by recipient cells can have profound functional impacts on the CNS. Thus, understanding how exosomes traverse the blood–brain barrier bidirectionally can have great therapeutic potential and diagnostic utility.
As the exosome field is witnessing exponential growth, it is perhaps an understatement to say that there is a requirement for more uniformity in exosome isolation and characterization methods. Refinement of exosome isolation in an in vivo setting will definitely enable the discovery of novel biological functions of exosomes. Many exosome studies have been performed using cells cultured in vitro. Future studies involving animal and clinical research will be a key to unlocking the potential of exosome biology. Particularly, a better understanding of the role played by exosomes in pathogenesis of neurodegeneration will pave the way for new therapeutic avenues. This is specifically significant as the aging population increases and with it a growing incidence of neurodegenerative diseases. The biological content of exosomes can be harnessed for biomarker discovery aiding in diagnosis and prognostic follow up studies. This is of particular importance as exosomes are present in most biological fluids and the biological cargo is stable and protected within the boundaries of exosome membranes.
References
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977.
- Morelli, A.E.; Larregina, A.T.; Shufesky, W.J.; Sullivan, M.L.G.; Stolz, D.B.; Papworth, G.D.; Zahorchak, A.F.; Logar, A.J.; Wang, Z.; Watkins, S.C.; et al. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood 2004, 104, 3257–3266.
- Bonifacino, J.S.; Rojas, R. Retrograde transport from endosomes to the trans-Golgi network. Nat. Rev. Mol. Cell Biol. 2006, 7, 568–579.
- Tu, Y.; Zhao, L.; Billadeau, D.D.; Jia, D. Endosome-to-TGN Trafficking: Organelle-Vesicle and Organelle-Organelle Interactions. Front. Cell Dev. Biol. 2020, 8, 163.
- Mullock, B.M.; Bright, N.A.; Fearon, C.W.; Gray, S.R.; Luzio, J.P. Fusion of Lysosomes with Late Endosomes Produces a Hybrid Organelle of Intermediate Density and Is NSF Dependent. J. Cell Biol. 1998, 140, 591–601.
- Bright, N.A.; Gratian, M.J.; Luzio, J. Endocytic Delivery to Lysosomes Mediated by Concurrent Fusion and Kissing Events in Living Cells. Curr. Biol. 2005, 15, 360–365.
- Grant, B.D.; Donaldson, J.G. Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2009, 10, 597–608.
- Abels, E.R.; Breakefield, X.O. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell. Mol. Neurobiol. 2016, 36, 301–312.
- Bebelman, M.P.; Bun, P.; Huveneers, S.; Van Niel, G.; Pegtel, D.M.; Verweij, F.J. Real-time imaging of multivesicular body–plasma membrane fusion to quantify exosome release from single cells. Nat. Protoc. 2020, 15, 102–121.
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500.
- Freeman, C.; Seaman, M.N.; Reid, E. The hereditary spastic paraplegia protein strumpellin: Characterisation in neurons and of the effect of disease mutations on WASH complex assembly and function. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2013, 1832, 160–173.
- Ropers, F.; Derivery, E.; Hu, H.; Garshasbi, M.; Karbasiyan, M.; Herold, M.; Nürnberg, G.; Ullmann, R.; Gautreau, A.; Sperling, K.; et al. Identification of a novel candidate gene for non-syndromic autosomal recessive intellectual disability: The WASH complex member SWIP. Hum. Mol. Genet. 2011, 20, 2585–2590.
- Courtland, J.L.; Bradshaw, T.W.; Waitt, G.; Soderblom, E.J.; Ho, T.; Rajab, A.; Vancini, R.; Kim, I.H.; Soderling, S.H. Genetic disruption of WASHC4 drives endo-lysosomal dysfunction and cognitive-movement impairments in mice and humans. eLife 2021, 10, e61590.
- Kilarski, W.; Jasiński, A. The formation of multivesicular bodies from the nuclear envelope. J. Cell Biol. 1970, 45, 205–211.
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002; Transport from the Trans Golgi Network to Lysosomes. Available online: https://www.ncbi.nlm.nih.gov/books/NBK26844/ (accessed on 6 January 2022).
- Piper, R.C.; Katzmann, D.J. Biogenesis and Function of Multivesicular Bodies. Annu. Rev. Cell Dev. Biol. 2007, 23, 519–547.
- Scott, C.; Vacca, F.; Gruenberg, J. Endosome maturation, transport and functions. Semin. Cell Dev. Biol. 2014, 31, 2–10.
- Peng, X.; Yang, L.; Ma, Y.; Li, Y.; Li, H. Focus on the morphogenesis, fate and the role in tumor progression of multivesicular bodies. Cell Commun. Signal. 2020, 18, 112.
- Matsuo, H.; Chevallier, J.; Mayran, N.; Le Blanc, I.; Ferguson, C.; Fauré, J.; Blanc, N.S.; Matile, S.; Dubochet, J.; Sadoul, R.; et al. Role of LBPA and Alix in Multivesicular Liposome Formation and Endosome Organization. Science 2004, 303, 531–534.
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brügger, B.; Simons, M. Ceramide Triggers Budding of Exosome Vesicles into Multivesicular Endosomes. Science 2008, 319, 1244–1247.
- Wei, D.; Zhan, W.; Gao, Y.; Huang, L.; Gong, R.; Wang, W.; Zhang, R.; Wu, Y.; Gao, S.; Kang, T. RAB31 marks and controls an ESCRT-independent exosome pathway. Cell Res. 2021, 31, 157–177.
- Clark, W.H. Electron Microscope Studies of Nuclear Extrusions in Pancreatic Acinar Cells of the Rat. J. Cell Biol. 1960, 7, 345–352.
- Andriana, B.B.; Mizukami, T.; Ishii, M.; Kanai, Y.; Kurohmaru, M.; Hayashi, Y. Postnatal development of multivesicular nuclear body in the Shiba goat Sertoli cell: An ultrastructural study. Okajimas Folia Anat. Jpn. 2004, 81, 15–24.
- Haines, H.; Baerwald, R.J. Nuclear membrane changes in herpes simplex virus-infected BHK-21 cells as seen by freeze-fracture. J. Virol. 1976, 17, 1038–1042.
- Conde-Vancells, J.; Rodriguez-Suarez, E.; Embade, N.; Gil, D.; Matthiesen, R.; Valle, M.; Elortza, F.; Lu, S.C.; Mato, J.M.; Falcon-Perez, J.M. Characterization and Comprehensive Proteome Profiling of Exosomes Secreted by Hepatocytes. J. Proteome Res. 2008, 7, 5157–5166.
- Kang, D.; Oh, S.; Ahn, S.-M.; Lee, B.-H.; Moon, M.H. Proteomic Analysis of Exosomes from Human Neural Stem Cells by Flow Field-Flow Fractionation and Nanoflow Liquid Chromatography−Tandem Mass Spectrometry. J. Proteome Res. 2008, 7, 3475–3480.
- Fauré, J.; Lachenal, G.; Court, M.; Hirrlinger, J.; Chatellard-Causse, C.; Blot, B.; Grange, J.; Schoehn, G.; Goldberg, Y.; Boyer, V.; et al. Exosomes are released by cultured cortical neurones. Mol. Cell. Neurosci. 2006, 31, 642–648.
- Sharma, P.; Mesci, P.; Carromeu, C.; McClatchy, D.R.; Schiapparelli, L.; Yates, J.R.; Muotri, A.R.; Cline, H.T. Exosomes regulate neurogenesis and circuit assembly. Proc. Natl. Acad. Sci. USA 2019, 116, 16086–16094.
- Chivet, M.; Javalet, C.; Laulagnier, K.; Blot, B.; Hemming, F.J.; Sadoul, R. Exosomes secreted by cortical neurons upon glutamatergic synapse activation specifically interact with neurons. J. Extracell. Vesicles 2014, 3, 24722.
- Bahrini, I.; Song, J.-H.; Diez, D.; Hanayama, R. Neuronal exosomes facilitate synaptic pruning by up-regulating complement factors in microglia. Sci. Rep. 2015, 5, 7989.
- Goldie, B.J.; Dun, M.; Lin, M.; Smith, N.D.; Verrills, N.; Dayas, C.; Cairns, M.J. Activity-associated miRNA are packaged in Map1b-enriched exosomes released from depolarized neurons. Nucleic Acids Res. 2014, 42, 9195–9208.
- Liu, H.-Y.; Huang, C.-M.; Hung, Y.-F.; Hsueh, Y.-P. The microRNAs Let7c and miR21 are recognized by neuronal Toll-like receptor 7 to restrict dendritic growth of neurons. Exp. Neurol. 2015, 269, 202–212.
- Lee, S.H.; Shin, S.M.; Zhong, P.; Kim, H.-T.; Kim, D.-I.; Kim, J.M.; Heo, W.D.; Kim, D.-W.; Yeo, C.-Y.; Kim, C.-H.; et al. Reciprocal control of excitatory synapse numbers by Wnt and Wnt inhibitor PRR7 secreted on exosomes. Nat. Commun. 2018, 9, 3434.
- Mederos, S.; González-Arias, C.; Perea, G. Astrocyte–Neuron Networks: A Multilane Highway of Signaling for Homeostatic Brain Function. Front. Synaptic Neurosci. 2018, 10, 45.
- Venturini, A.; Passalacqua, M.; Pelassa, S.; Pastorino, F.; Tedesco, M.; Cortese, K.; Gagliani, M.C.; Leo, G.; Maura, G.; Guidolin, D.; et al. Exosomes From Astrocyte Processes: Signaling to Neurons. Front. Pharmacol. 2019, 10, 1452.
- Abbott, N.J. Astrocyte-endothelial interactions and blood-brain barrier permeability. J. Anat. 2002, 200, 629–638.
- Sofroniew, M.V. Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 2015, 16, 249–263.
- Proia, P.; Schiera, G.; Mineo, M.; Ingrassia, A.M.R.; Santoro, G.; Savettieri, G.; Di Liegro, I. Astrocytes shed extracellular vesicles that contain fibroblast growth factor-2 and vascular endothelial growth factor. Int. J. Mol. Med. 2008, 21, 63–67.
- Peferoen, L.; Kipp, M.; Van Der Valk, P.; van Noort, J.; Amor, S. Oligodendrocyte-microglia cross-talk in the central nervous system. Immunology 2014, 141, 302–313.
- Jahn, O.; Siems, S.B.; Kusch, K.; Hesse, D.; Jung, R.B.; Liepold, T.; Uecker, M.; Sun, T.; Werner, H.B. The CNS Myelin Proteome: Deep Profile and Persistence After Post-mortem Delay. Front. Cell. Neurosci. 2020, 14, 239.
- Nave, K.-A. Myelination and the trophic support of long axons. Nat. Rev. Neurosci. 2010, 11, 275–283.
- Crain, J.M.; Nikodemova, M.; Watters, J.J. Microglia express distinct M1 and M2 phenotypic markers in the postnatal and adult central nervous system in male and female mice. J. Neurosci. Res. 2013, 91, 1143–1151.
- Potolicchio, I.; Carven, G.J.; Xu, X.; Stipp, C.; Riese, R.J.; Stern, L.J.; Santambrogio, L. Proteomic Analysis of Microglia-Derived Exosomes: Metabolic Role of the Aminopeptidase CD13 in Neuropeptide Catabolism. J. Immunol. 2005, 175, 2237–2243.
- Antonucci, F.; Turola, E.; Riganti, L.; Caleo, M.; Gabrielli, M.; Perrotta, C.; Novellino, L.; Clementi, E.; Giussani, P.C.; Viani, P.; et al. Microvesicles released from microglia stimulate synaptic activity via enhanced sphingolipid metabolism. EMBO J. 2012, 31, 1231–1240.
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608.
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11172–11177.
- Sharples, R.A.; Vella, L.J.; Nisbet, R.M.; Naylor, R.; Perez, K.; Barnham, K.J.; Masters, C.L.; Hill, A.F. Inhibition of γ-secretase causes increased secretion of amyloid precursor protein C-terminal fragments in association with exosomes. FASEB J. 2008, 22, 1469–1478.
- Walsh, D.M.; Selkoe, D.J. Deciphering the Molecular Basis of Memory Failure in Alzheimer’s Disease. Neuron 2004, 44, 181–193.
- Gouras, G.K.; Tampellini, D.; Takahashi, R.H.; Capetillo-Zarate, E. Intraneuronal β-amyloid accumulation and synapse pathology in Alzheimer’s disease. Acta Neuropathol. 2010, 119, 523–541.
- Sinha, M.S.; Ansell-Schultz, A.; Civitelli, L.; Hildesjö, C.; Larsson, M.; Lannfelt, L.; Ingelsson, M.; Hallbeck, M. Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol. 2018, 136, 41–56.
- Zheng, T.; Wu, X.; Wei, X.; Wang, M.; Zhang, B. The release and transmission of amyloid precursor protein via exosomes. Neurochem. Int. 2018, 114, 18–25.
- Winston, C.N.; Aulston, B.; Rockenstein, E.M.; Adame, A.; Prikhodko, O.; Dave, K.N.; Mishra, P.; Rissman, R.A.; Yuan, S.H. Neuronal Exosome-Derived Human Tau is Toxic to Recipient Mouse Neurons in vivo. J. Alzheimer’s Dis. 2019, 67, 541–553.
- Harding, C.; Heuser, J.; Stahl, P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J. Cell Biol. 1983, 97, 329–339.
- Pan, B.T.; Blostein, R.; Johnstone, R.M. Loss of the transferrin receptor during the maturation of sheep reticulocytes in vitro. An immunological approach. Biochem. J. 1983, 210, 37–47.
- Del Prete, D.; Rice, R.C.; Rajadhyaksha, A.M.; D’Adamio, L. Amyloid Precursor Protein (APP) May Act as a Substrate and a Recognition Unit for CRL4CRBN and Stub1 E3 Ligases Facilitating Ubiquitination of Proteins Involved in Presynaptic Functions and Neurodegeneration. J. Biol. Chem. 2016, 291, 17209–17227.
- Watanabe, T.; Hikichi, Y.; Willuweit, A.; Shintani, Y.; Horiguchi, T. FBL2 Regulates Amyloid Precursor Protein (APP) Metabolism by Promoting Ubiquitination-Dependent APP Degradation and Inhibition of APP Endocytosis. J. Neurosci. 2012, 32, 3352–3365.
- El Ayadi, A.; Stieren, E.S.; Barral, J.M.; Boehning, D. Ubiquilin-1 regulates amyloid precursor protein maturation and degradation by stimulating K63-linked polyubiquitination of lysine. Proc. Natl. Acad. Sci. USA 2012, 109, 13416–13421.
- Bustamante, H.A.; Rivera-Dictter, A.; Cavieres, V.A.; Muñoz, V.C.; González, A.; Lin, Y.; Mardones, G.A.; Burgos, P.V. Turnover of C99 is Controlled by a Crosstalk between ERAD and Ubiquitin-Independent Lysosomal Degradation in Human Neuroglioma Cells. PLoS ONE 2013, 8, e83096.
- Morel, E.; Chamoun, Z.; Lasiecka, Z.M.; Chan, R.B.; Williamson, R.L.; Vetanovetz, C.; Dall’Armi, C.; Simoes, S.; Du Jour, K.S.P.; McCabe, B.; et al. Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat. Commun. 2013, 4, 2250.
- Williamson, R.L.; Laulagnier, K.; Miranda, A.M.; Fernandez, M.A.; Wolfe, M.S.; Sadoul, R.; Di Paolo, G. Disruption of amyloid precursor protein ubiquitination selectively increases amyloid β (Aβ) 40 levels via presenilin 2-mediated cleavage. J. Biol. Chem. 2017, 292, 19873–19889.
- Xu, F.; Fu, Z.; Dass, S.; Kotarba, A.E.; Davis, J.; Smith, S.O.; Van Nostrand, W.E. Cerebral vascular amyloid seeds drive amyloid β-protein fibril assembly with a distinct anti-parallel structure. Nat. Commun. 2016, 7, 13527.
- An, K.; Klyubin, I.; Kim, Y.; Jung, J.H.; Mably, A.J.; O’Dowd, S.T.; Lynch, T.; Kanmert, D.; Lemere, C.A.; Finan, G.M.; et al. Exosomes neutralize synaptic-plasticity-disrupting activity of Aβ assemblies in vivo. Mol. Brain 2013, 6, 47.
- Dinkins, M.B.; Dasgupta, S.; Wang, G.; Zhu, G.; Bieberich, E. Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1792–1800.
- Yuyama, K.; Sun, H.; Usuki, S.; Sakai, S.; Hanamatsu, H.; Mioka, T.; Kimura, N.; Okada, M.; Tahara, H.; Furukawa, J.-I.; et al. A potential function for neuronal exosomes: Sequestering intracerebral amyloid-β peptide. FEBS Lett. 2014, 589, 84–88.
- Leverenz, J.B.; Quinn, J.F.; Zabetian, C.; Zhang, J.; Montine, K.S.; Montine, T.J. Cognitive impairment and dementia in patients with Parkinson disease. Curr. Top. Med. Chem. 2009, 9, 903–912.
- Weintraub, D.; Mamikonyan, E. The Neuropsychiatry of Parkinson Disease: A Perfect Storm. Am. J. Geriatr. Psychiatry 2019, 27, 998–1018.
- Borghi, R.; Marchese, R.; Negro, A.; Marinelli, L.; Forloni, G.; Zaccheo, D.; Abbruzzese, G.; Tabaton, M. Full length α-synuclein is present in cerebrospinal fluid from Parkinson’s disease and normal subjects. Neurosci. Lett. 2000, 287, 65–67.
- El-Agnaf, O.M.A.; Salem, S.A.; Paleologou, K.E.; Cooper, L.J.; Fullwood, N.J.; Gibson, M.J.; Curran, M.D.; Court, J.A.; Mann, D.M.A.; Ikeda, S.-I.; et al. α-Synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 2003, 17, 1–16.
- Howitt, J.; Hill, A.F. Exosomes in the Pathology of Neurodegenerative Diseases. J. Biol. Chem. 2016, 291, 26589–26597.
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-Produced alpha-Synuclein Is Secreted in a Calcium-Dependent Manner by Exosomes and Impacts Neuronal Survival. J. Neurosci. 2010, 30, 6838–6851.
- Tomlinson, P.R.; Zheng, Y.; Fischer, R.; Heidasch, R.; Gardiner, C.; Evetts, S.; Hu, M.; Wade-Martins, R.; Turner, M.R.; Morris, J.; et al. Identification of distinct circulating exosomes in Parkinson’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 353–361.
- Yang, Y.; Keene, C.; Peskind, E.R.; Galasko, D.R.; Hu, S.-C.; Cudaback, E.; Wilson, A.M.; Li, G.; Yu, C.-E.; Montine, K.S.; et al. Cerebrospinal Fluid Particles in Alzheimer Disease and Parkinson Disease. J. Neuropathol. Exp. Neurol. 2015, 74, 672–687.
- Alamri, Y.; Vogel, R.; Macaskill, M.; Anderson, T. Plasma exosome concentration may correlate with cognitive impairment in Parkinson’s disease. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2016, 4, 107–108.
- Grey, M.; Dunning, C.J.; Gaspar, R.; Grey, C.; Brundin, P.; Sparr, E.; Linse, S. Acceleration of α-Synuclein Aggregation by Exosomes. J. Biol. Chem. 2015, 290, 2969–2982.
- Kurzawa-Akanbi, M.; Tammireddy, S.; Fabrik, I.; Gliaudelytė, L.; Doherty, M.K.; Heap, R.; Matečko-Burmann, I.; Burmann, B.M.; Trost, M.; Lucocq, J.M.; et al. Altered ceramide metabolism is a feature in the extracellular vesicle-mediated spread of alpha-synuclein in Lewy body disorders. Acta Neuropathol. 2021, 142, 961–984.
- Surgucheva, I.; Sharov, V.S.; Surguchov, A. γ-Synuclein: Seeding of α-Synuclein Aggregation and Transmission between Cells. Biochem. 2012, 51, 4743–4754.
- Von Einem, B.; Eschbach, J.; Kiechle, M.; Wahler, A.; Thal, D.; McLean, P.; Weishaupt, J.H.; Ludolph, A.C.; Von Arnim, C.A.; Danzer, K.M. The Golgi-localized, gamma ear-containing, ARF-binding (GGA) protein family alters alpha synuclein (α-syn) oligomerization and secretion. Aging 2017, 9, 1677–1697.
- Stykel, M.G.; Humphries, K.M.; Kamski-Hennekam, E.; Buchner-Duby, B.; Porte-Trachsel, N.; Ryan, T.; Coackley, C.L.; Bamm, V.V.; Harauz, G.; Ryan, S.D. α-Synuclein mutation impairs processing of endomembrane compartments and promotes exocytosis and seeding of α-synuclein pathology. Cell Rep. 2021, 35, 109099.
- Chang, C.; Lang, H.; Geng, N.; Wang, J.; Li, N.; Wang, X. Exosomes of BV-2 cells induced by alpha-synuclein: Important mediator of neurodegeneration in PD. Neurosci. Lett. 2013, 548, 190–195.
- Xia, Y.; Zhang, G.; Kou, L.; Yin, S.; Han, C.; Hu, J.; Wan, F.; Sun, Y.; Wu, J.; Li, Y.; et al. Reactive microglia enhance the transmission of exosomal α-synuclein via toll-like receptor. Brain 2021, 144, 2024–2037.
- Bliederhaeuser, C.; Grozdanov, V.; Speidel, A.; Zondler, L.; Ruf, W.P.; Bayer, H.; Kiechle, M.; Feiler, M.S.; Freischmidt, A.; Brenner, D.; et al. Age-dependent defects of alpha-synuclein oligomer uptake in microglia and monocytes. Acta Neuropathol. 2015, 131, 379–391.
- Bosco, D.A.; Morfini, G.; Karabacak, N.M.; Song, Y.; Gros-Louis, F.; Pasinelli, P.; Goolsby, H.; Fontaine, B.A.; Lemay, N.; McKenna-Yasek, D.; et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 2010, 13, 1396–1403.
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133.
- Nonaka, T.; Masuda-Suzukake, M.; Arai, T.; Hasegawa, Y.; Akatsu, H.; Obi, T.; Yoshida, M.; Murayama, S.; Mann, D.M.; Akiyama, H.; et al. Prion-like Properties of Pathological TDP-43 Aggregates from Diseased Brains. Cell Rep. 2013, 4, 124–134.
- Grad, L.I.; Pokrishevsky, E.; Silverman, J.M.; Cashman, N.R. Exosome-dependent and independent mechanisms are involved in prion-like transmission of propagated Cu/Zn superoxide dismutase misfolding. Prion 2014, 8, 331–335.
- Basso, M.; Pozzi, S.; Tortarolo, M.; Fiordaliso, F.; Bisighini, C.; Pasetto, L.; Spaltro, G.; Lidonnici, D.; Gensano, F.; Battaglia, E.; et al. Mutant Copper-Zinc Superoxide Dismutase (SOD1) Induces Protein Secretion Pathway Alterations and Exosome Release in Astrocytes. J. Biol. Chem. 2013, 288, 15699–15711.
- Silverman, J.M.; Christy, D.; Shyu, C.C.; Moon, K.-M.; Fernando, S.; Gidden, Z.; Cowan, C.M.; Ban, Y.; Stacey, R.G.; Grad, L.I.; et al. CNS-derived extracellular vesicles from superoxide dismutase 1 (SOD1)G93A ALS mice originate from astrocytes and neurons and carry misfolded SOD1. J. Biol. Chem. 2019, 294, 3744–3759.
- Feneberg, E.; Steinacker, P.; Lehnert, S.; Schneider, A.; Walther, P.; Thal, D.; Linsenmeier, M.; Ludolph, A.C.; Otto, M. Limited role of free TDP-43 as a diagnostic tool in neurodegenerative diseases. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 351–356.
- Ding, X.; Ma, M.; Teng, J.; Teng, R.K.; Zhou, S.; Yin, J.; Fonkem, E.; Huang, J.; Wu, E.; Wang, X. Exposure to ALS-FTD-CSF generates TDP-43 aggregates in glioblastoma cells through exosomes and TNTs-like structure. Oncotarget 2015, 6, 24178–24191.
- Feiler, M.S.; Strobel, B.; Freischmidt, A.; Helferich, A.M.; Kappel, J.; Brewer, B.M.; Li, D.; Thal, D.; Walther, P.; Ludolph, A.C.; et al. TDP-43 is intercellularly transmitted across axon terminals. J. Cell Biol. 2015, 211, 897–911.
- Iguchi, Y.; Eid, L.; Parent, M.; Soucy, G.; Bareil, C.; Riku, Y.; Kawai, K.; Takagi, S.; Yoshida, M.; Katsuno, M.; et al. Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain 2016, 139, 3187–3201.
- Mustapic, M.; Eitan, E.; Werner, J.K., Jr.; Berkowitz, S.T.; Lazaropoulos, M.P.; Tran, J.; Goetzl, E.J.; Kapogiannis, D. Plasma Extracellular Vesicles Enriched for Neuronal Origin: A Potential Window into Brain Pathologic Processes. Front. Neurosci. 2017, 11, 278.
- Lai, C.P.; Mardini, O.; Ericsson, M.; Prabhakar, S.; Maguire, C.A.; Chen, J.W.; Tannous, B.A.; Breakefield, X.O. Dynamic Biodistribution of Extracellular Vesicles in Vivo Using a Multimodal Imaging Reporter. ACS Nano 2014, 8, 483–494.
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Mark, M.T.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335.
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Gardiner, C.; Sargent, I.L.; Wood, M.J.A.; Cooper, J.M. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367.

