Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jason Mercer | + 4280 word(s) | 4280 | 2022-02-24 03:21:19 | | | |
| 2 | Amina Yu | -170 word(s) | 4110 | 2022-03-04 03:59:32 | | | | |
| 3 | Amina Yu | -172 word(s) | 4108 | 2022-03-04 04:05:14 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Mercer, J. Vaccinia Virus Arrests and Shifts the Cell Cycle. Encyclopedia. Available online: https://encyclopedia.pub/entry/20186 (accessed on 25 June 2026).
Mercer J. Vaccinia Virus Arrests and Shifts the Cell Cycle. Encyclopedia. Available at: https://encyclopedia.pub/entry/20186. Accessed June 25, 2026.
Mercer, Jason. "Vaccinia Virus Arrests and Shifts the Cell Cycle" Encyclopedia, https://encyclopedia.pub/entry/20186 (accessed June 25, 2026).
Mercer, J. (2022, March 03). Vaccinia Virus Arrests and Shifts the Cell Cycle. In Encyclopedia. https://encyclopedia.pub/entry/20186
Mercer, Jason. "Vaccinia Virus Arrests and Shifts the Cell Cycle." Encyclopedia. Web. 03 March, 2022.
Copy Citation
Modulation of the host cell cycle is a common strategy used by viruses to create a pro-replicative environment. To facilitate viral genome replication, vaccinia virus (VACV) has been reported to alter cell cycle regulation and trigger the host cell DNA damage response. However, the cellular factors and viral effectors that mediate these changes remain unknown.
vaccinia virus
cell cycle
kinase
p53
DNA damage response
1. Introduction
Vaccinia virus (VACV), the smallpox vaccine and prototypic poxvirus are amongst the viruses shown to regulate the host cell cycle upon infection. Like all members of the Poxviridae, VACV is a large, double-stranded DNA virus which replicates exclusively in the cytoplasm of host cells [1][2]. To achieve this, VACV encodes and expresses a large subset of viral DNA replication proteins including a DNA polymerase, a helicase–primase, a uracil DNA glycosylase, a processivity factor, a single-stranded DNA-binding protein, a DNA ligase and the replicative protein kinase, B1 [3][4].
Despite its exclusively cytoplasmic replication, VACV has been shown to inhibit cell cycle progression and mitosis [5][6][7][8]. At a molecular level, infection was shown to alter expression of CDKs, cyclins, and the tumour suppressors Rb and p53 in pre-synchronised cells during late infection [9][10][11]. To date, the only VACV protein connected to cell cycle regulation is the viral kinase B1 [12]. Overexpression of B1 in the absence of infection was found to mediate hyperphosphorylation of p53, thereby promoting its degradation. B1-mediated degradation of p53 was shown to require the E3 ligase Mdm2, which is the negative regulator of p53, and the proteasome [12][13][14]. While B1-mediated degradation of p53 has not been investigated in the context of VACV infection, VACV was found to upregulate Mdm2 transcription, suggesting that a similar p53 degradation mechanism is employed during infection [11].
As a crucial effector in checkpoint signalling, p53 can pause the cell cycle and induce apoptosis. Cellular stress, such as DNA damage, promotes phosphorylation and stabilisation of p53 which then directs transcription of target genes, including the cell cycle inhibitor p21 that can arrest cells in G1/S and G2/M [15][16][17][18][19][20]. Conversely, inactivation of p53 prevents checkpoint-mediated cell cycle arrest in response to cellular stress, resulting in cell cycle dysregulation. Consistent with VACV-mediated dysregulation of the G1/S checkpoint through inhibition of p53 and Rb, a marked increase in S/G2 cells was observed during VACV infection [11].
Despite downregulation of these checkpoint effectors, VACV was shown to activate the cellular DNA damage response (DDR) to facilitate host protein-assisted viral genome replication [21]. The cellular DNA single-strand binding protein RPA was found to be recruited to replicating viral genomes, where it acts as a platform for replisome assembly. Within the replisome, the viral polymerase E9 was shown to interact with the DDR-activating protein topoisomerase II binding protein 1 (TOPBP1) and the cellular sliding clamp, proliferating cell nuclear antigen (PCNA). While activation of the DDR kinases ataxia telangiectasia and Rad3-related protein (ATR) and checkpoint kinase 1 (Chk1) was found to be essential, their role in VACV genome replication has not been established [21]. Additionally, mass spectrometry analysis of the proteins associated with replicating VACV genomes suggested that VACV might exploit several other cellular DNA repair pathways including Non-Homologous End Joining, Base Excision Repair, Nucleotide Excision Repair, Interstrand Crosslink Repair, and Homologous Recombination Repair (HRR) [22]. While neither ATR nor Chk1 was detected, PCNA was strongly enriched, as well as all subunits of the mini-chromosome maintenance MCM2-7 replicative helicase complex, the HRR components BLM, MRE11, NBS1, and Ku70, and topoisomerase I/II which was previously shown to facilitate VACV replication [23].
2. VACV Infection Inhibits Cell Proliferation
Cell proliferation is defined as the net change in cell number over time due to cell division and cell death. VACV infection has been reported to inhibit cell proliferation of mouse fibroblasts, and to have a pro-proliferative effect in human 143B osteosarcoma cells [11]. As this suggested that the effects of VACV infection on proliferation may be cell-type specific, it was asked how VACV infection impacted proliferation of HeLa, BSC40 and HCT116 cells. Mock-infected cells and cells infected with VACV were assessed at 0, 24 and 48 h post-infection (hpi) for cell number and cell death. While mock-infected cell numbers doubled every 24 h as expected, VACV-infected cell numbers did not increase over the 48 h time course in any of the cell lines (Figure 1A). The accompanying cytotoxicity assays showed that mock- and VACV-infected cells displayed the same level of cytotoxicity over 48 h, even when doubling the MOI (Figure 1B). These results indicated that VACV infection reduces cell proliferation through inhibition of cell division as opposed to induction of cell death.

Figure 1. VACV inhibits the host cell cycle. (A) HeLa, BSC40, and HCT116 cells were either mock infected, or infected with WT VACV at MOI 5 and cell numbers were determined over 48 h. (B) HeLa, BSC40, and HCT116 cells were either mock infected, or infected with WT VACV at MOI 5, and MOI 10. Cytotoxicity was measured using the Pierce LDH Cytotoxicity Assay Kit at 24 and 48 h and normalised to the cell lysis control. Data represent two biological replicates of technical triplicates and are displayed as the mean ± S.D. (C) VACV life cycle schematic highlighting the viral proteins L1 and D5 which are required for fusion and uncoating, respectively. The inactive viral fusion machinery is represented by red dots; its fusion-competent form by green dots. Black arrows indicate the respective next step in the virus life cycle. (D) BSC40 cells were either mock infected, or infected with VACV L1(+), or VACV L1(−) at MOI 5 and cell numbers were determined over 48 h. (E) BSC40 cells were either mock infected, or infected with VACV Cts24 at MOI 5 and incubated at either the permissive (31 °C), or non-permissive temperature (40 °C) and cell numbers were determined during 48 h. (A,D,E) Data represent biological triplicates, each with technical duplicates, and are displayed as the normalised mean ± S.E.M. Grey and black dots represent means of individual biological replicates.
3. VACV-Mediated Inhibition of Cell Proliferation Requires Virus Fusion but Not Genome Uncoating
As illustrated in Figure 1C, VACV enters host cells by macropinocytosis [24][25]. Once internalised, virions fuse from the macropinosome and deposit viral cores and their associated lateral bodies (LBs) into the host cytoplasm [26]. Pre-replicative early gene expression is initiated, allowing for the production of proteins required for subsequent genome uncoating [27], DNA replication and post-replicative intermediate and late gene expression [4]. Using recombinant VACVs defective for virus entry or genome uncoating, it was sought to define the stage of the virus life cycle required for inhibition of cell division. To test whether VACV entry was required, a recombinant VACV (vL1i) was used, which is inducible for the expression of the viral protein L1 [28]. The L1 protein is a member of the VACV entry–fusion complex, thus virions produced in the absence of L1, L1(−) virions are unable to mediate membrane fusion [28][29]. When BSC40 cells were infected with L1(+) virions, cell proliferation was inhibited as expected (Figure 1D). However, when cells were infected with L1(−) virions, they continued to multiply at a level similar to mock-infected cells, accumulating 2.7-fold over 48 h. This result indicated that a post-fusion cytoplasmic stage of the VACV lifecycle is required to inhibit cell proliferation.
4. VACV Infection Induces a General Cell Cycle Arrest
Having found that VACV inhibits cell proliferation, next it was asked at which cell cycle stage cells arrest upon infection. The cell cycle is divided into four consecutive phases: cell growth (G1), DNA replication (S), DNA repair (G2) and division (M). Cells that exit the cell cycle enter quiescence (G0), a dormant state marked by the absence of cell proliferation, CDK activity and reduced levels of the DNA replication licensing factor MCM2 [30][31][32][33][34][35].
Given the potent inhibition of cell proliferation observed upon VACV infection, first it was asked if VACV was driving cells into quiescence. When compared MCM2 protein levels between mock-infected control and VACV-infected cells over a 24 h time course (Figure 2A) it was found that infection did not significantly alter MCM2 levels compared to mock-infected cells (Figure 2B). This suggested that VACV does not drive cells into quiescence but rather inhibits an active stage of the cell cycle.
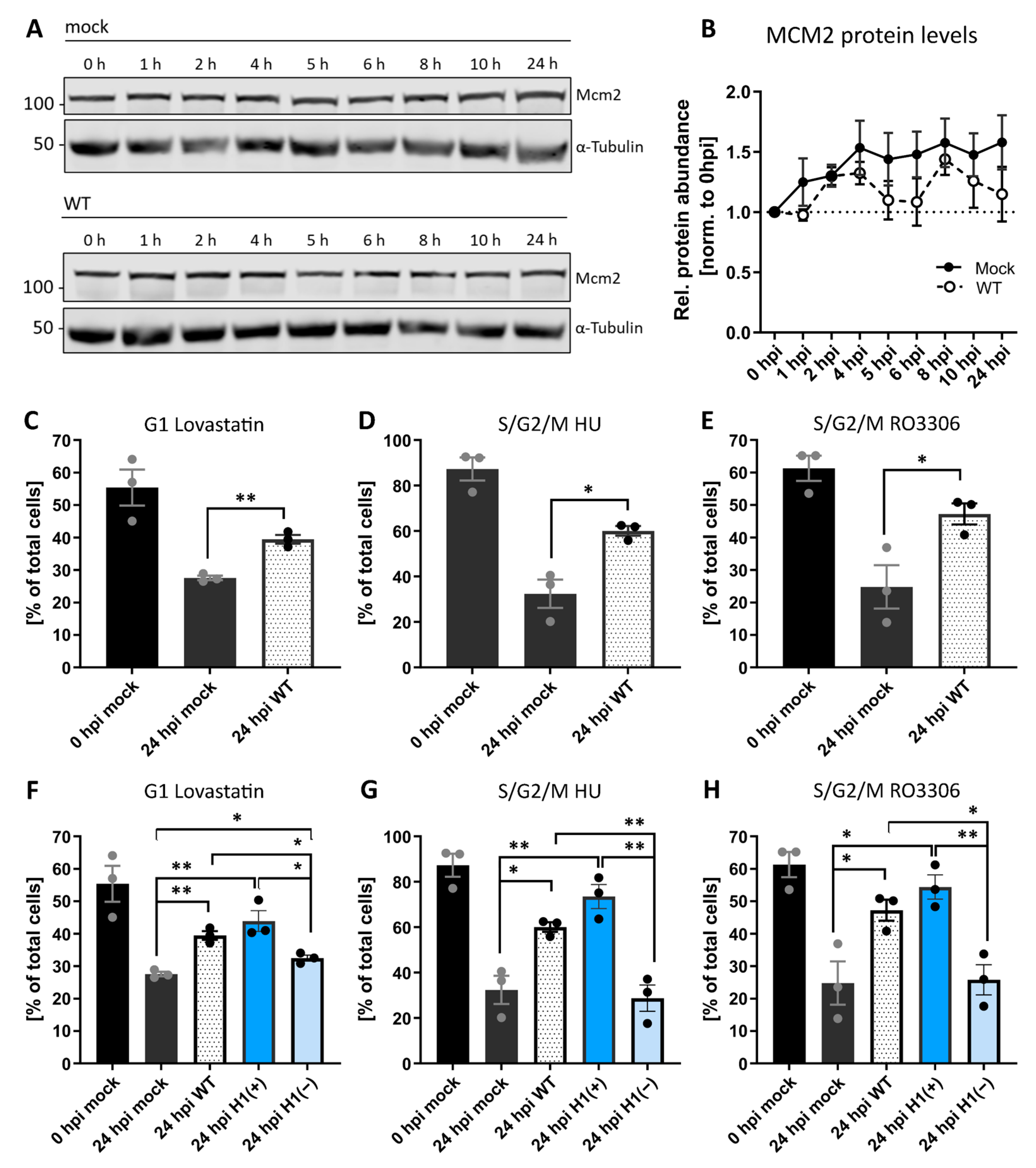
Figure 2. VACV establishes a general cycle block. (A) Immunoblot analysis of MCM2 during WT VACV infection. HeLa cells were either mock infected or infected with WT VACV at MOI 5 and harvested over 24 h. Whole-cell lysates were resolved via SDS-PAGE, and immunoblotted for MCM2, and α-Tubulin as loading control. A representative blot of three replicates is shown. (B) MCM2 protein abundance was quantified and normalised to the loading control. Data represent biological triplicates and are displayed as the mean ± S.E.M. [C–H] HeLa FUCCI cells were synchronised in the indicated cell cycle stage with either Lovastatin, HU, or the CDK1 inhibitor RO3306. Immediately after release, cells were mock infected (black), or infected with WT (white), H1(+) (blue), or H1(−) (light blue) VACV at MOI 2. The cell cycle distribution was assessed at 0, and 24 hpi by flow cytometry. (C,F) Percentage of G1 cells. [D–E, G–H] Percentage of S/G2/M cells. All data represent biological triplicates and are displayed as the mean ± S.E.M. Grey and black dots represent individual biological replicates. Data in (C–E) are replicated in (F–H). Parametric, unpaired, two-tailed t-test for significance. ns. p > 0.05, * p < 0.033, and ** p < 0.0021.
To identify which stage(s) were inhibited, it used HeLa FUCCI cells, which allowed to distinguish four different cell cycle phases based on fluorescence: early G1 (no fluorescence), G1 (red only), early S (red and green), and S/G2/M (green only) [36]. These cells were synchronised in either G1 with Lovastatin [37][38], S phase with hydroxyurea [39][40], or in late G2 with the CDK1 inhibitor RO3306 [41]. Cells were released, infected with VACV and the cell cycle distribution—relative to mock-infected cells—determined at 24 hpi. As expected, under all synchronisation and release conditions (Lovastatin, hydroxyurea and RO3306, all from Sigma-Aldrich, Gillingham, UK), mock-infected cells re-entered the cell cycle, resulting in reduced G1, S, and G2 fractions, respectively (Figure 2C–E; black bars). Conversely, a portion of cells infected with VACV were retained in the pre-synchronised stage independent of the synchronising agent: relative to mock-infected cells, 12% more VACV-infected cells remained in G1 after release from Lovastatin (Figure 2C), 28% more remained in S/G2/M after release from the hydroxyurea S-phase block (Figure 2D) and 22% more remained in S/G2/M after release from the G2 block mediated by RO3306 (Figure 2E). These results show that VACV inhibits G1, S, G2 and/or M-phase progression, suggesting that infection establishes a general cell cycle block.
It was found that a stage of the VACV lifecycle between fusion and uncoating inhibits cell proliferation (Figure 1), and viral early gene expression (EGE) occurs between these two, it was asked if EGE was required for the general block in cell cycle that was observed. To address this, a recombinant VACV were used, vindH1, which is inducible for the expression of the viral H1 phosphatase [42]. Virions produced in the absence of inducer (H1(−)) are attenuated for early gene expression [42][43]. As above, HeLa FUCCI cells were synchronised in G1, S or late G2, released and infected with H1(+) or H1(−) virions. Cells were then assessed for their cell cycle distribution relative to WT-infected cells. Cells infected with H1(+) virions, which are akin to WT virions, largely remained in their pre-synchronised cell cycle stage, while those infected with H1(−) virions were no longer capable of blocking cell cycle re-entry (Figure 2F–H). Relative to H1(+) virions, 11%, 45%, and 29% of cells re-entered the cell cycle after release from the G1, S or late G2 blocks, respectively. This indicates that the H1 phosphatase, a known immune modulator [26][44]—or viral EGE—is required for blocking cell cycle progression upon VACV infection.
4. VACV Infection Inhibits Cellular DNA Synthesis
It was found that VACV can retain cells in G1 and S/G2/M, the FUCCI system did not allow for further resolution of the individual effect of VACV on S, G2 and M. Therefore, whether VACV could inhibit M- and/or S-phase progression was addressed. To assess M phase during VACV infection, cells were infected with WT VACV, fixed, and stained with the DNA dye Hoechst at various time points between 0 and 24 hpi. Using genome condensation as a visual marker, it was quantified that the number of mitotic infected cells relative to the number of mitotic mock-infected cells at each time point (Figure 3A). While the percentage of mitotic cells fluctuated between 1.3% and 8.1%, the number of mitotic cells in infected samples declined until no mitotic cells were observed by 24 hpi (Figure 3A). This indicated that VACV is capable of blocking M phase either by preventing entry into mitosis, or by arresting cells prior to DNA condensation.
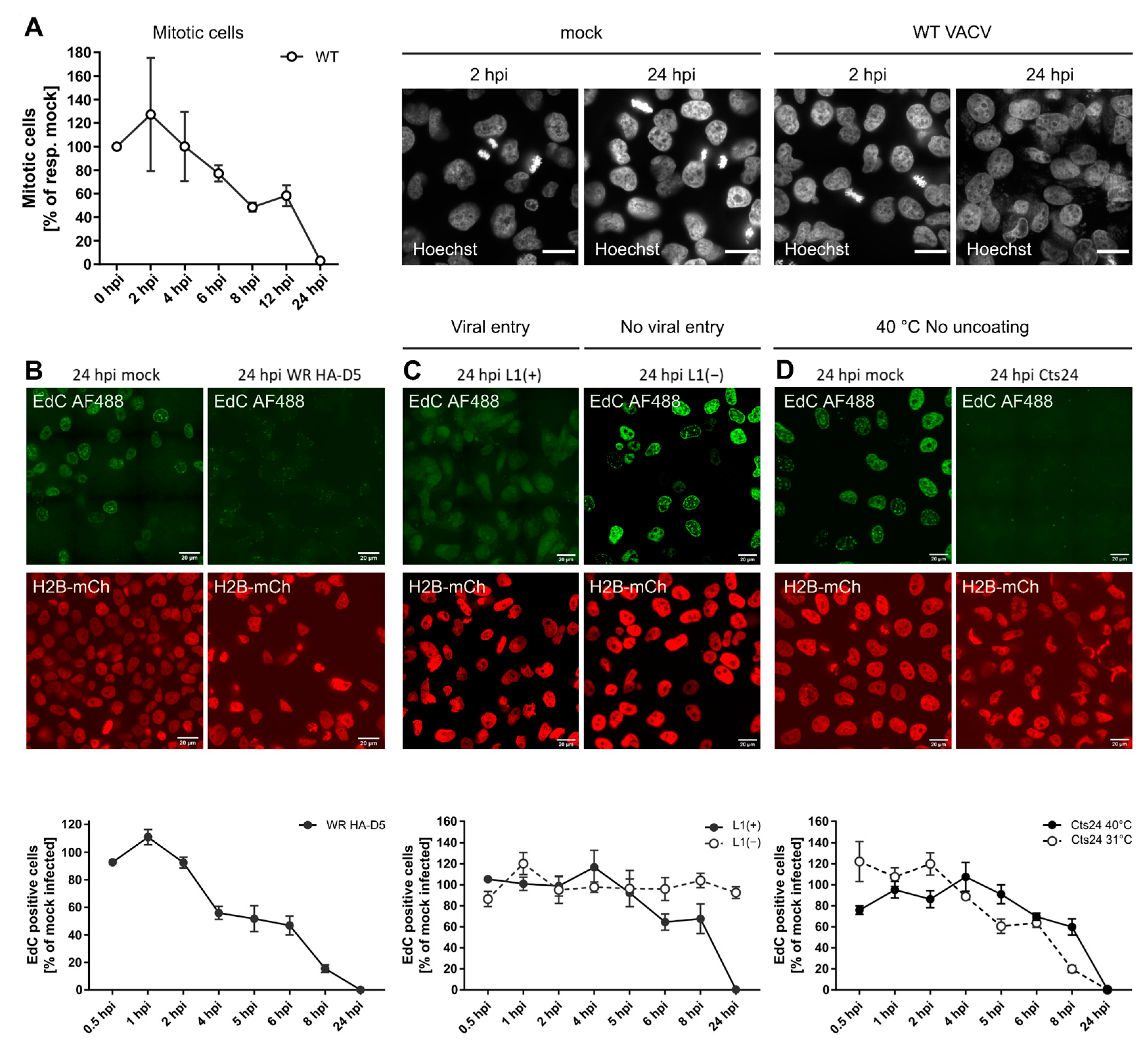
Figure 3. VACV inhibits mitosis and cellular DNA synthesis. (A) To quantify mitotic cells during VACV infection, HeLa cells were either mock infected, or infected with WT VACV at MOI 5, fixed and stained with the DNA dye Hoechst at various time points between 0 and 24 hpi. Using genome condensation as a visual marker, the number of mitotic infected cells was quantified relative to the number of mitotic mock-infected cells at each time point. Representative images are shown. Scale bar = 20 μm. Plotted data represent biological triplicates and are displayed as the mean ± S.E.M. (B–D) Pulse labelling with the nucleotide analogue EdC was used to identify active cellular DNA synthesis in VACV-infected cells. HeLa H2B-mCh cells were mock infected or infected with VACV at MOI 8 and pulse labelled with the nucleotide analogue EdC (10 μM) for 15 min prior to fixation. Incorporated EdC was detected using the Click-iTTM AF488 Imaging Kit and imaged by confocal microscopy. Representative micrographs are shown. Scale bar = 20μm. Per condition and biological replicate, 150–450 nuclei were scored either as EdC positive, or negative and normalised to the respective mock-infected control. Data represent biological triplicates and are displayed as the mean ± S.E.M. (B) BSC40 cells were infected with WT HA-D5 VACV. (C) The requirement for viral entry was tested by infecting BSC40 cells with recombinant VACV L1(+) and L1(−) VACV. (D) To test if viral entry was required, BSC40 cells were infected VACV Cts24 and incubated at either the permissive (31 °C), or non-permissive temperature (40 °C).
6. Inhibition of Cellular DNA Synthesis Is Independent of Viral Genome Replication
To determine which stage of the virus lifecycle was required for the observed inhibition of cellular DNA synthesis, viruses defective for either virus fusion (vL1i) or genome uncoating (Cts24) was described in Figure 1. It was found that fusion competent L1(+) virions abolished EdC incorporation by 24 hpi, while fusion-incompetent L1(−) virions appeared to have no impact on cellular DNA synthesis relative to controls (Figure 3C). As observed for inhibition of cell proliferation (Figure 1), a post-fusion cytoplasmic stage of the virus lifecycle is also responsible for inhibition of cellular DNA synthesis.
Using Cts24, it next asked if genome uncoating was required to inhibit cellular DNA synthesis. Infection under both permissive uncoating (Cts24 31 °C) and non-permissive uncoating (Cts24 40 °C) conditions resulted in complete inhibition of EdC incorporation by 24 hpi (Figure 3D). This indicated that neither viral genome uncoating, nor viral genome replication was required to block cellular DNA synthesis. Collectively, these results suggest that viral core deposition and/or a lateral body constituent are required to block cellular replication.
7. VACV Post-Replicative Gene Expression Shifts the Host Cell Cycle
The results indicate that VACV blocks host cell proliferation, arresting cells in G1, S, and G2, and inhibits both mitosis and DNA replication (Figure 1, Figure 2 and Figure 3). This suggested that VACV either inhibits the cell cycle systemically (i.e., upon infection cells remain in the cell cycle stage they are in), or that VACV arrests DNA replication and mitosis, thereby effectively trapping cells in either G1 or G2 phases of the cell cycle. To test this, it infected unsynchronised HeLa FUCCI cells with WT VACV and followed the cell cycle distribution over 24 h (Figure 4A). Relative to mock-infected cells, those infected with VACV gradually shifted from G1 into S/G2/M, resulting in a significant increase in the S/G2/M fraction at the expense of G1 by 24 hpi. Having established that infection inhibits cellular DNA replication (S phase) and mitosis (M phase), this result suggests that infected cells are shifted out of G1 and likely accumulate in S/G2.
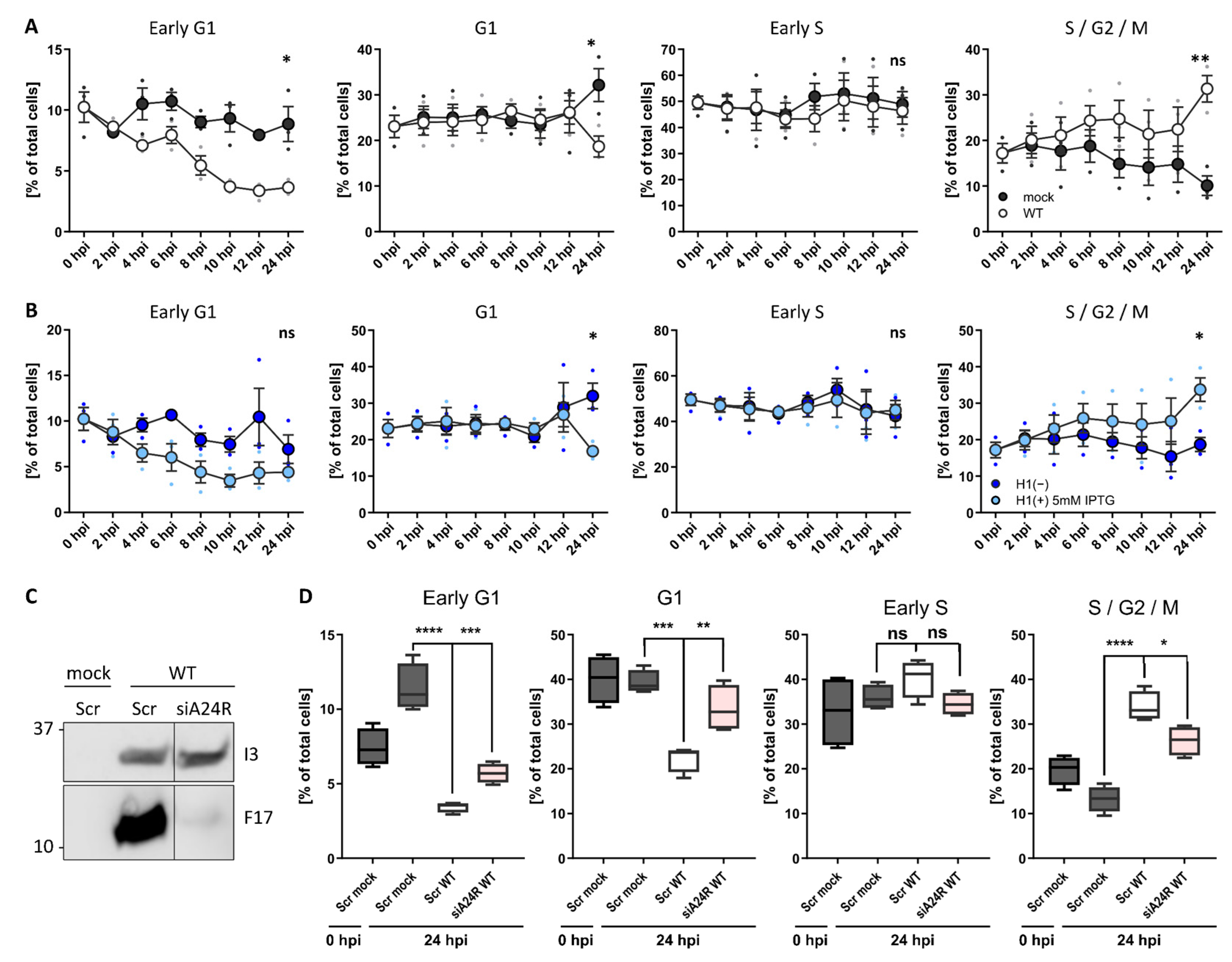
Figure 4. VACV post-replicative gene expression shifts the host cell cycle. (A) Cell cycle distribution of WT VACV-infected HeLa cells compared to uninfected controls. HeLa FUCCI cells were either mock infected or infected with WT VACV at MOI 2 and samples were harvested between 0 and 24 h. Cells were analysed by flow cytometry and the percentage G1, G1, early S, S/G2/M were assessed. Data represent biological triplicates and are displayed as the mean ± S.E.M. (B) Cell cycle distribution of VACV H1(−)-infected HeLa cells compared to parental H1(+)-infected controls. Samples were treated and analysed as in (A). Experiments represented in panel (A) and (B) were performed in parallel and share the same mock control. (C,D) To test if viral post-replicative gene expression was required to shift the cell cycle, the viral transcription factor A24 was silenced by siRNA. HeLa FUCCI cells were reverse transfected either with a scrambled control siRNA (Scr), or with siRNA targeting the viral transcription factor A24 (siA24R, rose). Samples were infected with WT VACV at MOI 1 and harvested at 24 hpi. (C) Inhibition of viral post-replicative gene expression was validated by immunoblot analysis of the viral early gene I3, and the viral late gene F17. (D) The cell cycle distribution was determined by flow cytometry and compared to Scr mock- (grey) and WT VACV (white)-infected control samples. Percentage of HeLa FUCCI cells in early G1, G1, early S, and S/G2/M. Data represent four biological replicates and are displayed as box (min to max), mean (line), and whiskers, indicating the 1 to 99 percentiles. Parametric, unpaired, two-tailed t-test for significance. ns. p > 0.05, * p < 0.033, ** p < 0.0021, *** p < 0.0002, and **** p < 0.0001.
To define which step of the virus life cycle was required to shift the host cell cycle out of G1, it tested the requirement for viral EGE by following the cell cycle distribution of unsynchronised HeLa FUCCI cells infected with H1(+) or H1(−) VACV (Figure 4B). As expected, H1(+)-infected cells were gradually shifted out of G1, resulting in accumulation in S/G2 by 24 hpi. In the absence of H1, VACV could no longer shift cells from G1 to S/G2, indicating that viral EGE and/or H1 was required to shift the host cell cycle. To narrow in, it was asked if post-replicative viral gene expression could mediate the cell cycle shift. For this, a component was depleted of the VACV DNA-dependent RNA polymerase, A24, using virus targeting siRNA [27]. As the VACV DNA-dependent RNA polymerase is pre-packaged in virions to direct in-core viral early gene transcription, siRNA depletion of A24 only affects post-replicative intermediate and late gene transcription which rely on newly expressed A24. The effectiveness of this approach was demonstrated by monitoring the expression of an early (I3) and late gene (F17) after A24 depletion (Figure 4C). Relative to mock-infected cells treated with control siRNA, WT VACV infection had shifted the distribution of cells from G1 into S/G2 by 24 h as expected (Figure 4D; Scr WT). Depletion of A24 prevented the shift from G1 to S/G2 in WT-infected samples (Figure 4D; siA24R WT). Compared to scrambled WT VACV controls, the G1 fraction was significantly increased, and the S/G2 fraction significantly decreased when viral post-replicative gene expression was inhibited. These findings suggest that viral post-replicative gene expression is required to shift the host cell cycle from G1 to S/G2 late in infection.
8. The Viral Kinase F10 Activates the Cellular DDR Late in Infection
To gain additional insight into how VACV arrests and shifts the host cell cycle, it was turned to the underlying cell signalling pathways that control these processes. Cyclin-dependent kinases (CDKs) and their activators, the cyclins, are the key drivers of cell cycle progression. Periodic expression of cyclins targets the activity of their partner CDK to defined phases of the cell cycle. It has been shown that VACV alters CDK and cyclin expression in synchronised rabbit fibroblasts, as well as the transcript levels of cyclins in unsynchronised HeLa cells [9][11]. To test if VACV altered the expression of CDKs and cyclins at the protein level in unsynchronised HeLa cells, it analysed CDK 1, 2, 4, 6, 7 and cyclins A, B, D, and E by immunoblot analysis over 24 h. Contrary to previous findings with synchronised cells [10], neither CDK nor cyclin protein levels were found to be differentially regulated during the first 24 h of infection.
As these results indicate that VACV does not prevent proliferation by depleting positive regulators, it next focused on cell cycle inhibitors. Molecular checkpoints can either pause the cell cycle or induce apoptosis upon aberrant cell cycle events such as DNA damage. The kinases ATR and/or ATM detect DNA breaks and trigger the cellular DNA damage response (DDR) through phosphorylation of the kinases Chk1 and/or Chk2 [45][46][47]. As VACV has been reported to activate the DDR in a pre-uncoating step [21], it asked if DDR was the trigger for cell cycle arrest during early infection. First, it monitored DDR activation during VACV infection using phosphorylation of Chk1 (Ser345) and Chk2 (Thr68) as a readout. Immunoblot analysis of WT VACV-infected HeLa cells showed Chk1 Ser345 phosphorylation after 8 hpi, and Chk2 Thr68 phosphorylation from 4 hpi, with a strong signal increase after 6 hpi (Figure 5A,B). DDR activation did not occur in mock controls, and was not due to cellular DNA damage as determined by immunoblots directed against phosphorylation of γH2AX Ser139 (Figure 5A). While these findings confirmed robust DDR activation in response to VACV infection, the kinetics of the response under the experimental setting was not consistent with a pre-uncoating event as the trigger [21].
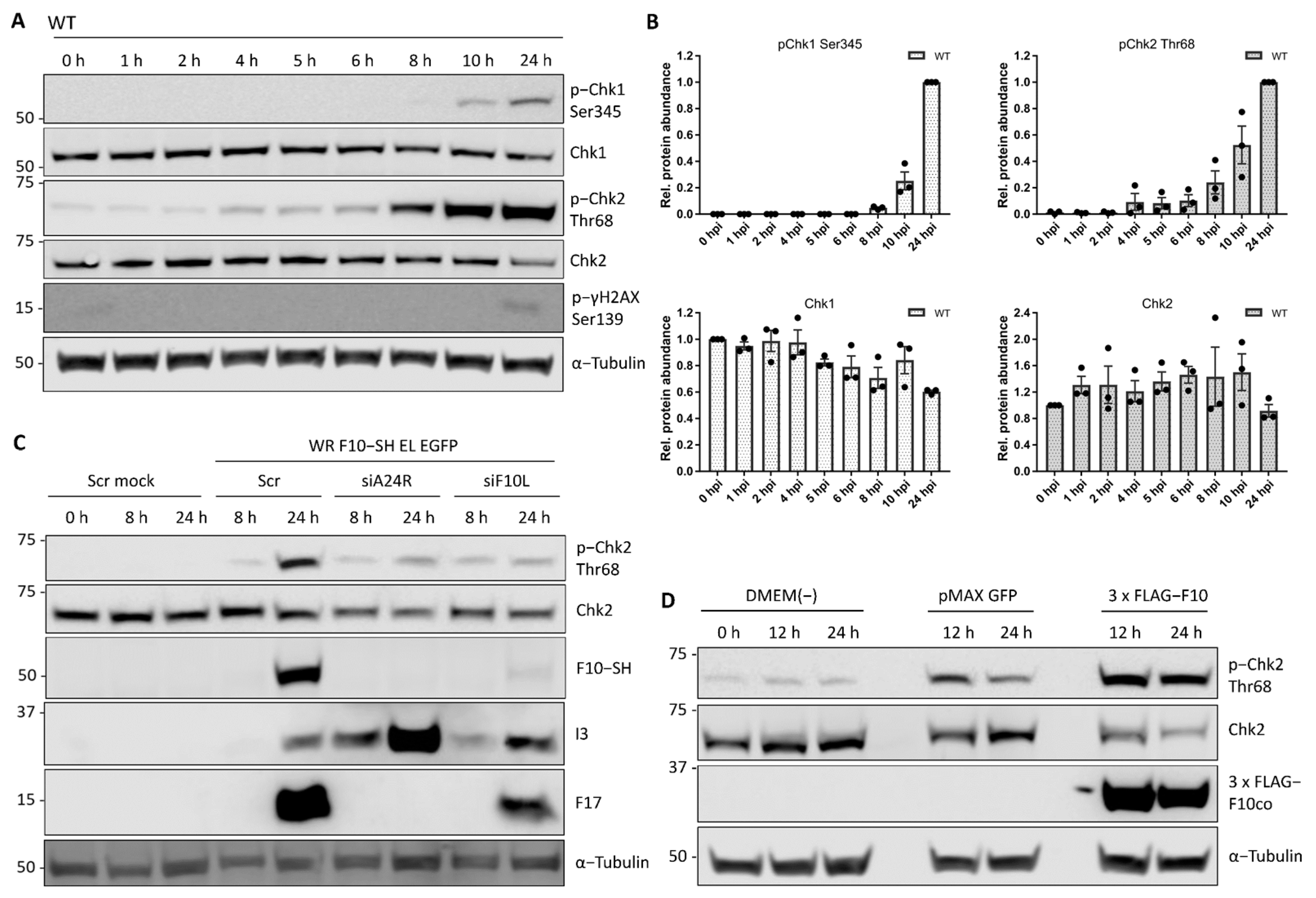
Figure 5. The viral kinase F10 activates the cellular DNA damage response. DDR activation was measured by phosphorylation of the DDR effector kinases Chk1 and Chk2, respectively. (A) HeLa cells were infected with WT VACV at MOI 5 and samples were harvested between 0 and 24 h. Whole-cell lysates were resolved via SDS-PAGE and immunoblotted for activating phosphorylation of Chk1 (Ser345), Chk1, activating phosphorylation of Chk2 (Thr 68), phosphorylated γH2AX (Ser139) which marks cellular DNA damage, and α-Tubulin as loading control. A representative blot of biological triplicates is shown. (B) Total and phosphorylated protein abundance was quantified and normalised to the loading control. Data represent biological triplicates, normalised to 0 h, and 24 h, respectively, and are displayed as the mean ± S.E.M. Black dots represent individual biological replicates. (C) The late viral kinase F10 is required to activate the cellular DDR. HeLa cells were reverse transfected with scrambled control siRNA (Scr), siA24R, or siF10L. At 48 h after transfection, cells were either mock infected, or infected with WR F10-SH EL EGFP (MOI 1) and samples were harvested at 0, 8, and 24 hpi. Whole-cell lysates were resolved via SDS-PAGE and immunoblotted for pChk2 (Thr68), Chk2, HA to detect expression of viral F10-SH, the early viral protein I3, the late viral protein F17, and α-Tubulin as loading control. A representative blot is shown. (D) The late viral kinase F10 is sufficient to activate the cellular DDR. HeLa cells were transfected with either a DMEM(-) control, pMAX GFP control vector, or codon-optimised 3 × FLAG-F10co. Samples were harvested at 0 h, 12 h, and 24 h post-transfection. Whole-cell lysates were resolved via SDS-PAGE and immunoblotted for activating phosphorylation of Chk2 (Chk2 Thr68), Chk2, FLAG to monitor expression of 3 × FLAG-F10co, and α-Tubulin as loading control. A representative blot of three biological replicates is shown.
The late phosphorylation of Chk1 and Chk2 suggested that a post-replicative event in the VACV life cycle activated the DDR. It therefore tested the requirement for viral intermediate and late gene expression by silencing A24 (siA24R). HeLa cells were infected with a recombinant VACV strain, WR F10-SH EL EGFP, that expresses a C-terminal streptavidin-HA tagged version of F10 from its endogenous locus and EGFP under a viral Early/Late promoter from the TK locus. While the scrambled control siRNA did not affect VACV-mediated Chk2 activation, siA24R strongly reduced Chk2 Thr68 phosphorylation in infected cells (Figure 5C). This implicated a viral intermediate/late protein in Chk2 activation. As activation is driven by phosphorylation, it was hypothesised that the late VACV-encoded kinase, F10 [48][49], may be responsible. F10 was depleted using siRNA (siF10L) and the knockdown confirmed by immunoblot directed against the SH tag (Figure 5C; F10-SH). It was found that depletion of F10 prevented Chk2 Thr68 phosphorylation, suggesting that F10 is required for DDR activation.
These results show that VACV F10 is sufficient to trigger phosphorylation and activation of the DDR kinase Chk2 in the absence of infection.
References
- Condit, R.C.; Moussatche, N.; Traktman, P. In A Nutshell: Structure and Assembly of the Vaccinia Virion. In Advances in Virus Research; Academic Press: Cambridge, MA, USA, 2006; Volume 66, pp. 31–124.
- Dales, S.; Siminovitch, L. The development of vaccinia virus in Earle’s L strain cells as examined by electron microscopy. J. Biophys. Biochem. Cytol. 1961, 10, 475–503.
- Evans, E.; Traktman, P. Characterization of vaccinia virus DNA replication mutants with lesions in the D5 gene. Chromosoma 1992, 102, S72–S82.
- Moss, B. Poxviridae: The viruses and their replication. In Fields Virology; Knipe, D., Howley, P., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 2007; Volume 74, p. 2906.
- Jungwirth, C.; Launer, J. Effect of Poxvirus Infection on Host Cell Deoxyribonucleic Acid Synthesis. J. Virol. 1968, 2, 401–408.
- Kit, S.; Dubbs, D.R. Biochemistry of vaccinia-infected mouse fibroblasts (strain L-M): I. Effects on nuclei acid and protein synthesis. Virology 1962, 18, 274–285.
- Magee, W.E.; Sagik, B.P. The synthesis of deoxyribonucleic acid by HeLa cells infected with vaccinia virus. Virology 1959, 8, 134–137.
- Magee, W.E.; Sheek, M.R.; Burrous, M.J. The synthesis of vaccinial deoxyribonucleic acid. Virology 1960, 11, 296–299.
- Guerra, S.; Lopez-Fernandez, L.A.; Pascual-Montano, A.; Munoz, M.; Harshman, K.; Esteban, M. Cellular Gene Expression Survey of Vaccinia Virus Infection of Human HeLa Cells. J. Virol. 2003, 77, 6493–6506.
- Wali, A.; Strayer, D.S. Infection with Vaccinia Virus Alters Regulation of Cell Cycle Progression. DNA Cell Biol. 1999, 18, 837–843.
- Yoo, N.-K.; Pyo, C.-W.; Kim, Y.; Ahn, B.-Y.; Choi, S.-Y. Vaccinia virus-mediated cell cycle alteration involves inactivation of tumour suppressors associated with Brf1 and TBP. Cell. Microbiol. 2008, 10, 583–592.
- Santos, C.R.; Vega, F.M.; Blanco, S.; Barcia, R.; Lazo, P.A. The vaccinia virus B1R kinase induces p53 downregulation by an Mdm2-dependent mechanism. Virology 2004, 328, 254–265.
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299.
- Kubbutat, M.H.G.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303.
- Agarwal, M.L.; Agarwal, A.; Taylor, W.R.; Stark, G.R. p53 controls both the G2/M and the G1 cell cycle checkpoints and mediates reversible growth arrest in human fibroblasts. Proc. Natl. Acad. Sci. USA 1995, 92, 8493–8497.
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512.
- Dornan, D.; Shimizu, H.; Burch, L.; Smith, A.J.; Hupp, T.R. The proline repeat domain of p53 binds directly to the transcriptional coactivator p300 and allosterically controls DNA-dependent acetylation of p53. Mol. Cell. Biol. 2003, 23, 8846–8861.
- Kastan, M.B.; Zhan, Q.; El-Deiry, W.S.; Carrier, F.; Jacks, T.; Walsh, W.V.; Plunkett, B.S.; Vogelstein, B.; Fornace, A.J. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 1992, 71, 587–597.
- Taylor, W.R.; Stark, G.R. Regulation of the G2/M transition by p53. Oncogene 2001, 20, 1803–1815.
- Waldman, T.; Kinzler, K.W.; Vogelstein, B. p21 Is Necessary for the p53-mediated G1 Arrest in Human Cancer Cells. Cancer Res 1995, 55, 5187–5190.
- Postigo, A.; Ramsden, A.E.; Howell, M.; Way, M. Cytoplasmic ATR Activation Promotes Vaccinia Virus Genome Replication. Cell Rep. 2017, 19, 1022–1032.
- Reyes, E.D.; Kulej, K.; Pancholi, N.J.; Akhtar, L.N.; Avgousti, D.C.; Kim, E.T.; Bricker, D.K.; Spruce, L.A.; Koniski, S.A.; Seeholzer, S.H.; et al. Identifying Host Factors Associated with DNA Replicated During Virus Infection. Mol. Cell Proteom. 2017, 16, 2079–2097.
- Lin, Y.-C.J.; Li, J.; Irwin, C.R.; Jenkins, H.; DeLange, L.; Evans, D.H. Vaccinia Virus DNA Ligase Recruits Cellular Topoisomerase II to Sites of Viral Replication and Assembly. J. Virol. 2008, 82, 5922–5932.
- Mercer, J.; Helenius, A. Vaccinia Virus Uses Macropinocytosis and Apoptotic Mimicry to Enter Host Cells. Science 2008, 320, 531–535.
- Chang, S.-J.; Chang, Y.-X.; Izmailyan, R.; Tang, Y.-L.; Chang, W. Vaccinia Virus A25 and A26 Proteins Are Fusion Suppressors for Mature Virions and Determine Strain-Specific Virus Entry Pathways into HeLa, CHO-K1, and L Cells. J. Virol. 2010, 84, 8422–8432.
- Schmidt, F.I.; Bleck, C.K.E.; Reh, L.; Novy, K.; Wollscheid, B.; Helenius, A.; Stahlberg, H.; Mercer, J. Vaccinia Virus Entry Is Followed by Core Activation and Proteasome-Mediated Release of the Immunomodulatory Effector VH1 from Lateral Bodies. Cell Rep. 2013, 4, 464–476.
- Kilcher, S.; Schmidt, F.I.; Schneider, C.; Kopf, M.; Helenius, A.; Mercer, J. siRNA Screen of Early Poxvirus Genes Identifies the AAA+ ATPase D5 as the Virus Genome-Uncoating Factor. Cell Host Microbe. 2014, 15, 103–112.
- Bisht, H.; Weisberg, A.S.; Moss, B. Vaccinia Virus L1 Protein Is Required for Cell Entry and Membrane Fusion. J. Virol. 2008, 82, 8687–8694.
- Gray, R.D.M.; Albrecht, D.; Beerli, C.; Huttunen, M.; Cohen, G.H.; White, I.J.; Burden, J.J.; Henriques, R.; Mercer, J. Nanoscale polarization of the entry fusion complex of vaccinia virus drives efficient fusion. Nat. Microbiol. 2019, 4, 1636–1644.
- Coller, H.A.; Sang, L.; Roberts, J.M. A new description of cellular quiescence. PLoS Biol. 2006, 4, e83.
- Mlcochova, P.; Sutherland, K.A.; Watters, S.A.; Bertoli, C.; de Bruin, R.A.; Rehwinkel, J.; Neil, S.J.; Lenzi, G.M.; Kim, B.; Khwaja, A.; et al. A G1-like state allows HIV-1 to bypass SAMHD1 restriction in macrophages. EMBO J. 2017, 36, 604–616.
- Musahl, C.; Holthoff, H.P.; Lesch, R.; Knippers, R. Stability of the Replicative Mcm3 Protein in Proliferating and Differentiating Human Cells. Exp. Cell Res. 1998, 241, 260–264.
- So, W.-K.; Cheung, T.H. Molecular Regulation of Cellular Quiescence: A Perspective from Adult Stem Cells and Its Niches. In Cellular Quiescence: Methods and Protocols; Lacorazza, H.D., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2018; pp. 1–25. ISBN 978-1-4939-7371-2.
- Terzi, M.Y.; Izmirli, M.; Gogebakan, B. The cell fate: Senescence or quiescence. Mol. Biol. Rep. 2016, 43, 1213–1220.
- Tsuruga, H.; Yabuta, N.; Hashizume, K.; Ikeda, M.; Endo, Y.; Nojima, H. Expression, Nuclear Localization and Interactions of Human MCM/P1 Proteins. Biochem. Biophys. Res. Commun. 1997, 236, 118–125.
- Sakaue-Sawano, A.; Kurokawa, H.; Morimura, T.; Hanyu, A.; Hama, H.; Osawa, H.; Kashiwagi, S.; Fukami, K.; Miyata, T.; Miyoshi, H.; et al. Visualizing Spatiotemporal Dynamics of Multicellular Cell-Cycle Progression. Cell 2008, 132, 487–498.
- Gray-Bablin, J.; Rao, S.; Keyomarsi, K. Lovastatin Induction of Cyclin-dependent Kinase Inhibitors in Human Breast Cells Occurs in a Cell Cycle-independent Fashion. Cancer Res. 1997, 57, 604–609.
- Rao, S.; Porter, D.C.; Chen, X.; Herliczek, T.; Lowe, M.; Keyomarsi, K. Lovastatin-mediated G1 arrest is through inhibition of the proteasome, independent of hydroxymethyl glutaryl-CoA reductase. Proc. Natl. Acad. Sci. USA 1999, 96, 7797–7802.
- Bacchetti, S.; Whitmore, G.F. The Action of Hydroxyurea on Mouse L-Cells*. Cell Prolif. 1969, 2, 193–211.
- Fallon, R.J.; Cox, R.P. Cell cycle analysis of sodium butyrate and hydroxyurea, inducers of ectopic hormone production in HeLa cells. J. Cell. Physiol. 1979, 100, 251–261.
- Vassilev, L.T.; Tovar, C.; Chen, S.; Knezevic, D.; Zhao, X.; Sun, H.; Heimbrook, D.C.; Chen, L. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc. Natl. Acad. Sci. USA 2006, 103, 10660–10665.
- Liu, K.; Lemon, B.; Traktman, P. The dual-specificity phosphatase encoded by vaccinia virus, VH1, is essential for viral transcription in vivo and in vitro. J. Virol. 1995, 69, 7823–7834.
- Novy, K.; Kilcher, S.; Omasits, U.; Bleck, C.K.E.; Beerli, C.; Vowinckel, J.; Martin, C.K.; Syedbasha, M.; Maiolica, A.; White, I.; et al. Proteotype profiling unmasks a viral signalling network essential for poxvirus assembly and transcriptional competence. Nat. Microbiol. 2018, 3, 588.
- Najarro, P.; Traktman, P.; Lewis, J.A. Vaccinia Virus Blocks Gamma Interferon Signal Transduction: Viral VH1 Phosphatase Reverses Stat1 Activation. J. Virol. 2001, 75, 3185–3196.
- Awasthi, P.; Foiani, M.; Kumar, A. ATM and ATR signaling at a glance. J. Cell Sci. 2015, 128, 4255–4262.
- Maréchal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect Biol. 2013, 5, a012716.
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204.
- Lin, S.; Broyles, S.S. Vaccinia protein kinase 2: A second essential serine/threonine protein kinase encoded by vaccinia virus. Proc. Natl. Acad. Sci. USA 1994, 91, 7653–7657.
- Punjabi, A.; Traktman, P. Cell biological and functional characterization of the vaccinia virus F10 kinase: Implications for the mechanism of virion morphogenesis. J. Virol. 2005, 79, 2171–2190.
More
Information
Subjects:
Virology; Cell Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Revisions:
3 times
(View History)
Update Date:
04 Mar 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No