 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Laurens Maertens | + 8011 word(s) | 8011 | 2020-09-09 08:45:55 | | | |
| 2 | Rita Xu | -4353 word(s) | 3658 | 2020-09-14 04:16:50 | | |
Video Upload Options
Bacteria are increasingly used for biotechnological applications such as bioremediation, biorecovery, bioproduction, and biosensing. The development of strains suited for such applications requires a thorough understanding of their behavior, with a key role for their transcriptomic landscape. We present a thorough analysis of the transcriptome of Cupriavidus metallidurans CH34 cells acutely exposed to copper by tagRNA-sequencing. C. metallidurans CH34 is a model organism for metal resistance, and its potential as a biosensor and candidate for metal bioremediation has been demonstrated in multiple studies. In total, 7500 transcription start sites (TSSs) were annotated and classified with respect to their location relative to coding sequences (CDSs). Predicted TSSs were used to re-annotate 182 CDSs. The TSSs of 2422 CDSs were detected, and consensus promotor logos were derived. Interestingly, many leaderless messenger RNAs (mRNAs) were found. In addition, many mRNAs were transcribed from multiple alternative TSSs. We observed pervasive intragenic TSSs both in sense and antisense to CDSs. Antisense transcripts were enriched near the 50 end of mRNAs, indicating a functional role in post-transcriptional regulation. In total, 578 TSSs were detected in intergenic regions, of which 35 were identified as putative small regulatory RNAs. Finally, we provide a detailed analysis of the main copper resistance clusters in CH34, which include many intragenic and antisense transcripts. These results clearly highlight the ubiquity of noncoding transcripts in the CH34 transcriptome, many of which are putatively involved in the regulation of metal resistance.
1. Introduction
Environmental pollution with toxic metals due to anthropogenic activities is an internationally growing concern [1][2][3]. The exposure and the risk of elevated concentrations of these pollutants in the environment can lead to bioaccumulation and harmful effects [4][5][6][7], which are facilitated by the high toxicity and recalcitrance of some metals [8][9]. Consequently, monitoring tools for metal accumulation in natural environments such as soils and water bodies are needed. Physicochemical analysis techniques, while accurate and sensitive, often fail to chart the bioavailability and the toxicity of the polluting components [10][11][12]. At the same time, microorganisms show great promise as biosensors to quantify the bioavailable fraction of heavy metals such as copper [13], an essential trace element that is highly toxic when overly abundant [14]. In addition, microorganisms can be used to combat environmental contamination with heavy metals in a process called bioremediation [3][8][15][16]. Bioremediation has been the focus of extensive research in recent years as a clean and efficient alternative to conventional strategies (reviewed in Tabak et al., 2005 [17] and Akcil et al., 2015 [18]).
Cupriavidus metallidurans strains are exemplary β-proteobacteria in metal-contaminated industrial environments [19]. Type strain CH34 was first isolated from a decantation basin in a zinc factory near Engis, Belgium, in 1976 [14][20]. It was quickly shown to encode resistance mechanisms to a wide range of metals [21] and has become a model organism to study heavy metal resistance (HMR) in bacteria [19]. Copper resistance in strain CH34 is mediated by multiple cooperating Cu detoxification systems; copF and copA1B1C1D1 encode a Cu efflux P-type ATPase and a periplasmic detoxification system, respectively, and are part of the extensive 21-gene cop cluster. The neighboring silDCBA cluster encodes a heavy metal efflux- resistance nodulation division (HME-RND) driven system. These gene clusters are encoded on the pMOL30 megaplasmid, and homologous systems can be found on its chromosome and chromid (Table 1). In addition, many more accessory genes may play a role in Cu resistance. The integration of these systems brings about a minimum inhibitory concentration of 3 mM Cu2+, three times higher than that of Escherichia coli K38 [22][23]. A detailed description of Cu resistance in strain CH34 can be found in Mergeay and Van Houdt [19].
Table 1. Overview of the main Cu resistance gene clusters in Cupriavidus metallidurans CH34.
| Gene Cluster | Locus Tag 1 | Replicon 2 | Homologous Gene Cluster | Locus Tag 1 | Replicon 2 | Function |
|---|---|---|---|---|---|---|
| copA1B1C1D1 | 6112–6115 | pMOL30 | copA2B2C2D2 | 5671–5668 | CHR2 | Periplasmic detoxification |
| copF | 6119 | pMOL30 | cupA | 3524 | CHR1 | PIB1-type ATPase |
| silCBA | 6133–6136 | pMOL30 | cusCBA | 5031–5033 | CHR2 | HME-RND efflux pump |
In a previous study, biosensors for metals such as Pb2+, Zn2+, and Cu2+ have been developed based on Cupriavidus strains [24][25]. These biosensors function via a transcriptional fusion of metal-specific promotor regions to a luxCDABE luciferase gene cluster, which allows for the emission of a measurable bioluminescent signal upon translation (BIOMET® system). The strength of this signal is proportional to the biologically available fraction of a specific metal. Cupriavidus-based biosensors have been used for the characterization of bioavailable metal fractions in soil, sediments, mineral wastes, etc. [26][27][28]. In addition, the bioremediation potential of CH34, owing to its capacity for metal solubilization and biocrystallization, has been demonstrated in several experimental setups [24][29]. For instance, the deposition of ZnCO3 and CdCO3 crystals around the cell surface, both biologically catalyzed and via abiotic processes, can lead to efficient depletion of these toxic metal ions from the environment. Cupriavidus strains are especially interesting in the case of mixed pollution with metals and recalcitrant organic compounds, since they are often able to degrade a broad array of aromatic compounds [30][31]. However, the construction of bacterial strains able to efficiently cope with mixed pollutions remains challenging [32].
In order to understand, optimize, and control the molecular processes underlying the above mentioned applications, it is vital to understand their regulation. Indeed, an elaborate network of sigma factors [33][34] and transcriptional regulators [35][36] has been found to play an important role in CH34. However, new paradigms in bacterial gene regulation highlight the role of small regulatory RNAs (sRNAs), acting at a (post-)transcriptional level in disparate stress responses [37][38][39]. For instance, sRNAs are involved in a plethora of biological processes, including virulence [40][41][42][43][44][45][46][47], antibiotic resistance [48][49][50][51][52] and mobile genetic elements [53] as well as oxidative and metal stress [54][55][56][57][58][59][60] and the degradation of organic compounds [61].
Thus, in order to fully understand the intricate response of C. metallidurans CH34, a highly detailed map of its transcriptome is a basic necessity. Although its genome is fully sequenced and annotated [31], and transcriptome data in response to metal stress are available [22][34], these data do not allow analyzing pertinent features such as transcription start sites (TSSs), 5′ untranslated regions (5′ UTRs), RNA processing sites (PSSs) and regulatory RNAs. Therefore, we used a tagRNA-sequencing (tagRNA-seq) approach, a modified RNA-seq method that is based on the differential labeling of 5’ RNA ends and that enables whole transcriptome sequencing with the discrimination of primary from processed 5’ RNA ends [62][63]. The ability of tagRNA-seq to annotate TSSs has been well established in recent years, and it is commonly exploited in research on regulatory features of the bacterial transcriptome. Copper was used as imposed stress, since CH34′s copper resistance mechanism is unique in respect to its complexity compared to others [19], and the putative functions of sRNAs in the bacterial copper response remain insufficiently studied.
2. Transcriptional Start Site Profiling
2.1. General Characteristics of Detected TSSs
In a next step, the tagRNA-seq data were used to identify and probe the type of the 5′ ends of RNAs in order to obtain a global snapshot of the transcriptional organization in C. metallidurans CH34 and to scrutinize the impact of Cu stress on the RNA landscape. The transcription start site (TSS) detection algorithm, as described in Materials and Methods, was fine-tuned to annotate the 7500 most highly expressed TSSs in both the control and the copper condition. This number was roughly based on the number of annotated genes and pseudogenes in CH34 (6514). These TSSs were divided into primary, secondary, internal, antisense, and orphan TSS according to their location, similar to previous publications [64] with minor modifications. A primary TSS (pTSS) is the main TSS of a gene or operon and is located within 200 bp upstream of a start codon. It is expressed at least twice as strongly as the second most highly expressed TSS within those 200 bps. The remaining TSSs in this region were classified as secondary TSSs (sTSSs). An internal TSS (iTSS) is located within and on the coding strand, while an antisense TSSs (aTSS) is located on the non-coding strand of a CDS or within 100 bp upstream of its start codon. The orphan TSSs (oTSSs) are not associated with CDSs. An overview of this classification for every replicon is shown in Table 2. As TSS classification was prioritized via the following cascade: pTSS > sTSS > iTSS > aTSS > oTSS; there is no overlap between the different classes. In addition, the tagRNA-seq data was verified with 5′ and 3′ RACE of selected transcripts, including copA1 messenger RNA (mRNA) and several antisense and orphan transcripts (see below). Similarly, the previously identified TSSs of cnrC, czcI, and pbrA were also detected and confirmed by the tagRNA-seq results [65][66][67], both of which corroborate the validity of the TSS identification with the tagRNA-seq procedure.
Table 2. Number and overlap of detected transcription start sites (TSSs) in each replicon 1.
| pTSS | sTSS | iTSS | aTSS | oTSS | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CT | Cu | ∩ | CT | Cu | ∩ | CT | Cu | ∩ | CT | Cu | ∩ | CT | Cu | ∩ | |
| CHR1 | 1490 | 1382 | 1231 | 729 | 734 | 483 | 1294 | 1390 | 916 | 1015 | 934 | 723 | 278 | 248 | 216 |
| CHR2 | 741 | 674 | 586 | 187 | 166 | 102 | 497 | 526 | 359 | 499 | 446 | 348 | 161 | 155 | 134 |
| pMOL28 | 45 | 43 | 38 | 19 | 19 | 13 | 89 | 83 | 74 | 97 | 92 | 77 | 21 | 22 | 18 |
| pMOL30 | 86 | 108 | 78 | 17 | 42 | 11 | 100 | 150 | 76 | 129 | 132 | 101 | 53 | 54 | 46 |
| Genome | 2362 | 2207 | 1933 | 952 | 961 | 609 | 1980 | 2149 | 1425 | 1740 | 1638 | 1249 | 513 | 479 | 414 |
2.2. Primary TSSs
Primary TSSs were detected for 2422 CDSs, amounting to 35.8% of all annotated CDSs. In a first step, this information was combined with the output of the recently developed GeneMarkS-2 gene prediction tool [68]. CDSs for which no pTSS was detected and that contained an iTSS between the original start codon and the newly predicted one, being preceded by a predicted ribosome-binding site (RBS), were scrutinized. This resulted in the putative reannotation of 182 CDSs. In addition, the consensus RBS and the spacer length were derived (Figure 2).
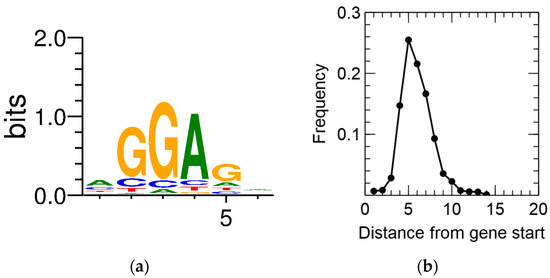
Figure 2. Ribosome-binding site sequence motif (a) and spacer length distribution (b) of C. metallidurans CH34.
Four cases were related to metal resistance. First, related to copper, copM encoding a 136 aa uncharacterized pre-protein was putatively re-annotated to be 114 aa. The cleavage site was similar for both, i.e., the processed proteins were identical, but the signal peptide prediction was much better for the newly annotated CDS (0.82 vs. 0.66; SignalP-5.0). In addition, cupC encoding 133 aa Cu chaperone was putatively reannotated to be 66 aa. Related to the czc cluster involved in cadmium, zinc, and cobalt resistance, czcN (273 aa; 30.1 kDa predicted molecular weight) encoding a membrane-bound isoprenylcysteine carboxyl methyltransferase was putatively reannotated to be 216 aa (23.7 kDa predicted molecular weight). CzcN is homologous to NccN of Alcaligenes xylosoxidans 31 (reclassified as C. metallidurans), which was found to be a 23.5 kDA protein [69]. Finally, the pbrD gene coding for a Pb2+-binding protein [66] could be putatively reannotated based on both TSS identification and GeneMarkS-2 prediction (CDS and RBS). However, the new CDS is not in frame and codes for a 150 aa protein unrelated to PbrD. These results merit further study of its function and sequence, since pbrD appears not to be necessary for Pb2+ resistance and is absent in all other known lead-resistant bacteria [70]. Moreover, although it is heterologously expressed in E. coli, it does show Pb2+ absorption [71].
Next, the 5′ untranslated region (5′ UTR) lengths and their distribution were calculated (Figure 3), which showed a peak at a length of 26 nt, after which it tapered off. No difference was noted between the control and the Cu condition, indicating that Cu exposure does not entail a global measurable bias in the 5′ UTR length of expressed genes. A curious observation was the high number of leaderless mRNA (4.67% of all pTSSs), defined as those mRNAs with a 5′ UTR length shorter than 5 nt. Because of the lack of a 5′ UTR, these transcripts are translated via non-canonical mechanisms. Similar observations have been made in Burkholderia cenocepacia [72], Streptomyces coelicor [73], Salmonella enterica [74], Caulobacter crescentus [75][76], and Helicobacter pylori [77][78]. A functional enrichment analysis showed that leaderless mRNAs was significantly enriched in eggNOG class K (transcription), which has already been observed in many bacterial species [79], and class L (replication, recombination, and repair). Several leaderless mRNAs coding for transcriptional regulators were also differentially expressed in the presence of Cu2+, including arsR, ompR, ohrR, and zniR. In addition, copJ, merP, rpoN, and czcI2 were transcribed into leaderless mRNAs. The prevalence of leaderless mRNAs has been shown to be higher in organisms with an earlier evolutionary age, and has been linked to extreme habitats [79][80].
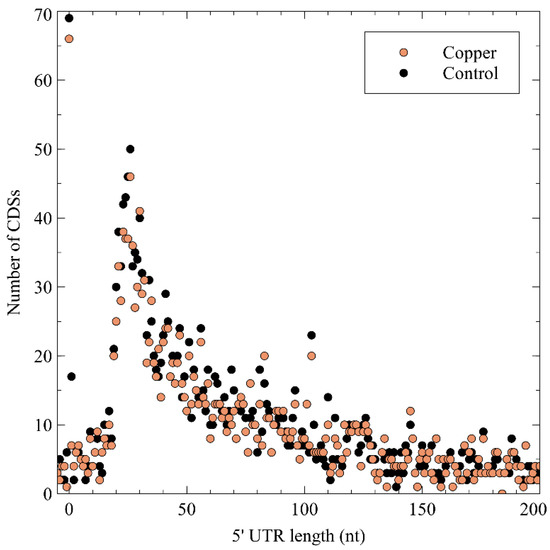
Figure 3. The 5′ untranslated region (UTR) length distribution in the C. metallidurans transcriptomes. CDS: coding sequence.
Finally, to complete the promoter information, the consensus sequence in a region of 100 nt around each pTSS was identified with Improbizer [81], which allowed us to weight the location of detected motifs. On the chromosome, the chromid, and the megaplasmid pMOL30, a TAnAAT consensus motif was detected around the −10 position, generally flanked by GC-rich stretches. At the −35 position, a TTGACA-like motif with higher variability was detected, again flanked by short GC-rich stretches. It is possible that CDSs preceded by these consensus motifs, which are similar in location and sequence to those found in E. coli, are under transcriptional control of the two housekeeping σ70-factors rpoD1 (Rmet_2606) and rpoD2 (Rmet_4661) [31]. Interestingly, although pMOL28 and pMOL30 do not contain essential housekeeping genes, TAnAAT consensus motifs were found on pMOL30 but not pMOL28, indicating a difference in sigma factor recruitment for the initiation of pMOL28 and pMOL30 gene transcription. Noteworthy, in contrast with pMOL30, which does not encode any sigma factors, pMOL28 encodes the ECF sigma factor CnrH. However, it is only involved in transcription of the cnr locus [65]. In addition, it has been shown that none of the remaining 10 ECF sigma factors, which are encoded on the chromosome (6) and the chromid (4), are necessary for the upregulation of any of the operons responding to metal shock [34]. Nevertheless, the involvement of non-plasmidic sigma factors in the transcription of plasmidic genes is not unexpected. On pMOL30, most CDSs preceded by a TAnAAT motif have an unknown function, but some genes involved in metal resistance are preceded by a TAnAAT-like pattern (czcE, pbrT, ubiE, and copM). Further determination of the functions of the remaining hypothetical proteins on both pMOL28 and pMOL30 may provide conclusive insights in this matter.
2.3. Secondary TSSs
Alternative mRNAs deriving from secondary TSSs were detected for 623 CDSs. Among the genes differentially expressed under Cu stress, a high number of regulatory proteins was readily apparent, including the main regulators of pMOL30-based (copR1) and chromid-based periplasmic (copR2) Cu detoxification systems as well as ohrR (Rmet_3619), zniS (Rmet_5322), bzdR (Rmet_1223), iscR, rpoH, and czcR2. In addition, several genes involved in HMR and stress responses were transcribed from alternative TSSs. Alternative TSSs have been reported in several bacterial and archaeal species [82][83][84] and have been linked to differences in translational efficiency and mRNA stability [85]. Longer 5′ UTRs enable more extensive post-transcriptional regulation, e.g., via sRNAs, and can contain riboswitches that respond to various environmental cues [86]. Interestingly, many mRNAs coding for sigma factors showed multiple TSSs, including rpoA, rpoD1, rpoE, rpoH, rpoI, rpoK, and rpoM, which could have an impact on the transcription of genes associated with a particular sigma factor.
2.4. Intragenic TSSs
The second most abundant TSS class after the pTSSs was that of the iTSSs. As all TSSs were derived from primary 5′-PPP-RNA, these iTSSs are unlikely to be the result of RNA degradation, e.g., by 5′-exonucleases, although some could be pTSSs or sTSSs due to misannotated CDSs, or transcriptional noise from spurious promotors. More importantly, iTSSs can also mark the start site of noncoding RNAs.
Even though the roles of the detected iTSSs are unclear, they are pervasive in the CH34 transcriptome. Functional enrichment analysis showed that, in the copper condition, eggNOG classes C, F, J, M, O, P, and T were enriched in iTSSs. In total, 55 of the 167 genes involved in metal resistance contained at least 1 iTSS, e.g., almost all cop1 genes and follow-up functional analyses could provide more insights into their specific role in metal resistance. The iTSSs related to the Cu resistance mechanisms (cop1 and sil clusters on pMOL30) are provided and discussed in detail below.
In both the control and the Cu condition, iTSSs were enriched in the first 10% of the encompassing CDS, while iTSSs were relatively depleted in the last 10% (Figure 4). This observation lends credibility to the hypothesis that many detected iTSSs are pTSSs or sTSSs to misannotated CDSs. However, these were not included in our re-annotation, as the current approach does not allow discriminating between the original and the putative new start codon. Of course, these uncertainties could be addressed with additional protein-based experiments such as proteome profiling (although not straightforward as the absence of a longer protein over the presence of a shorter one needs to be shown), but this is out of the scope of this study. Since there is currently no evidence that iTSSs in the first percentile are linked more to coding RNAs than in other percentiles, we conclude that many iTSSs either mark the start site of noncoding RNA or are the result of transcriptional noise.
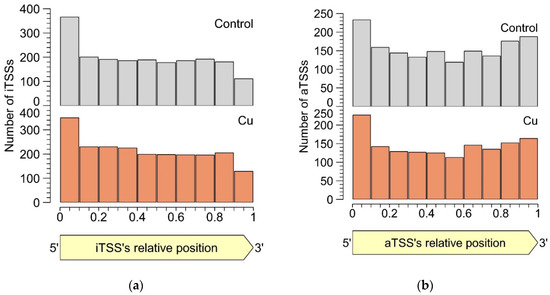
Figure 4. Percentile-wise distribution of iTSSs (a) and aTSSs (b) positions relative to the cognate CDS.
2.5. Antisense TSSs
Similar to intergenic TSSs, antisense TSSs are ubiquitously found in the CH34 transcriptome. In total, 2129 aTSSs were associated with 692 CDSs, indicating that many mRNAs can putatively be complexed by multiple antisense transcripts. Of all aTSSs, 408 showed a logFC smaller than −1 or greater than 1, indicating differential expression. Six differentially expressed aTSSs were validated by 5′ RACE experiments. It is generally assumed that aTSSs mark the start of noncoding transcripts, however, whether a detected antisense transcript has a (regulatory) function or is the result of transcriptional noise [87] needs to be determined case-by-case. Since antisense transcripts can obscure regulatory features of the sense transcript by perfect base pairing [88], the location of aTSSs relative to the 5′ and 3′ ends of the sense transcript was scrutinized (Figure 4b). There is a clear enrichment of aTSSs near the 5′ end of the sense CDS, with a less prominent enrichment near the 3′ end, indicating a putative regulatory role of the antisense transcripts.
Specifically related to copper resistance, aTSSs were found in silB, copA1, copF, copG, copH, copL, and copM (Figure 5 and Figure 6). The aTSSs in silB (nt position 168,587 on pMOL30) and copL (nt position 181,548 on pMOL30) were further selected for 3′ RACE validation, indicating transcript lengths of approximately 700 and 500 bp, respectively. In addition, an aTSS was found in czcA1.
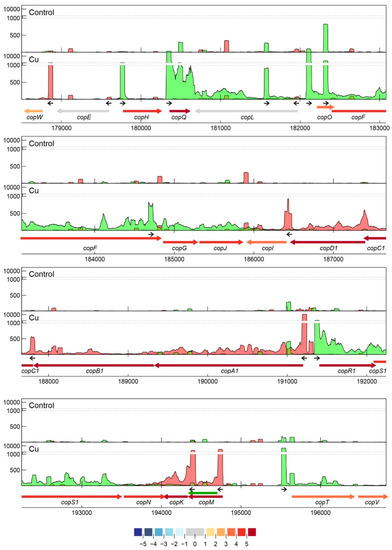
Figure 5. Transcription profile analysis of the cop cluster from C. metallidurans CH34 when exposed to copper. Combined TSS read counts of the three biological replicates for control (upper) and Cu condition (lower) are shown for positive (green) and negative (red) strands, with the y-axis containing a break pair (1000–1200) represented as striped grey lines. CDSs related to the cop cluster (coordinates for the pMOL30 region shown at the bottom) are colored based on their log2 fold change. The small black arrows indicate clearly identified primary and internal TSSs. The green arrow represents a re-annotated CDS.
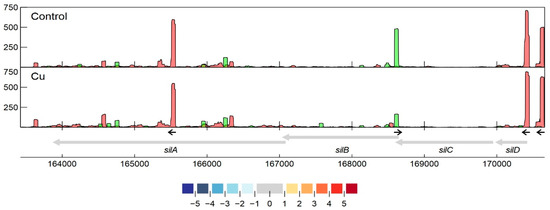
Figure 6. Transcription profile analysis of the sil cluster from C. metallidurans CH34 when exposed to copper. Combined TSS read counts of the three biological replicates for control (upper) and Cu condition (lower) are shown for positive (green) and negative (red) strands. CDSs related to the sil cluster (coordinates for the pMOL30 region shown at the bottom) are colored based on their log2 fold change. The small black arrows indicate clearly identified primary and internal TSSs.
Antisense TSSs were enriched in CDSs belonging to eggNOG classes L (replication, recombination, and repair), M (cell wall/membrane/envelope biogenesis), and T (signal transduction mechanisms) in the control condition, and classes J (translation, ribosomal structure and biogenesis), L, and M in the copper condition. Class T was only significantly overrepresented in the control condition, which could indicate that repression of translation via antisense transcription is common for CDSs related to signal transduction and partially relieved under copper stress. Many sigma factors, such as rpoB (Rmet_3334), rpoD1 (Rmet_2606), rpoN, rpoB, and rpoS (Rmet_2115), showed antisense transcription to a high extent. The role of alternative sigma factors in the initiation of antisense transcription has been described before [89][90][91]; inversely, an interesting case of the regulation of the sigma factor RpoS itself by antisense and anti-antisense RNAs has been shown in E. coli [92].
2.6. Orphan TSSs
In total, 578 TSSs were detected in intergenic regions unassociated with CDSs. These orphan TSSs could mark the start of transcripts resulting from the 5′ of mRNAs with exceptionally long 5′ UTRs, unidentified CDSs, often coding for short peptides [93], and trans-acting sRNAs [94], or they could result from transcription by spurious promoters.
First, the prevalence of long 5′ UTRs was investigated. A 200–300 nt region upstream of known start codons was scrutinized for oTSSs, and uninterrupted coverage from the oTSS to the start codon was manually verified. In this way, 126 oTSSs marked the 5′ of putative long 5′ UTRs. Interestingly, hfq, coding for a small RNA-binding protein that stabilizes sRNAs and modulates RNA–RNA interactions [95] showed a long 5′ UTR (209 bp) next to two other mRNA isoforms with 5′ UTRs of 25 and 137 bp. Relative expressions of these three isoforms (25, 137, and 209 nt 5′ UTR) were 6.1%, 81.1%, and 12.8% and 15.0%, 74.7%, and 10.3% for control and copper conditions, respectively. These disparate 5′ UTRs might have given rise to differences in post-transcriptional regulation of the hfq mRNA. While the relative coverage of each alternative TSS was quite similar in control and copper conditions, it is possible that stronger selection of differential TSSs exists in other (stress) conditions.
In a subsequent analysis, the existence of putative open reading frames (ORFs) downstream of oTSSs was studied. In total, 417 oTSSs were located upstream an ORF with ATG, GTG, or TTG as start codons and either with a minimum length of 150 nt or with homology to proteins in the non-redundant protein database. A list of these proteins with their inferred function, among which are several transporters, can be found in sheet “oTSS ORF BLAST”.
Consequently, the remaining 35 oTSS were neither located within 300 nt upstream of known CDSs nor associated with putative ORFs. These oTSSs were assumed to mark the 5′ end of noncoding RNAs. Eleven oTSSs showed a log2 fold change above 1 or below −1. As the expression of these oTSSs is affected by Cu stress, they are especially interesting as candidates for sRNAs with regulatory functions in the response to metal exposure. Three of them, based on interesting locations relative to known HMR genes, were experimentally validated by 5′ RACE. Two of these differentially expressed orphan transcripts were also experimentally validated by 3′ RACE (one at CHR1 nt position 3,784,309 and one at CHR2 nt position 147,562), indicating lengths of roughly 550 bp and 450 bp. It must be mentioned that 3′ ends of coding and non-coding RNAs cannot be accurately inferred from tagRNA-seq coverage, hence the need for additional experimental validation (via 3′ RACE or other techniques).
References
- Valette-Silver, N.J. The Use of Sediment Cores to Reconstruct Historical Trends in Contamination of Estuarine and Coastal Sediments. Estuaries 1993, 16, 577–588, doi:10.2307/1352796.
- Nicholson, F.A.; Smith, S.; Alloway, B.; Carlton-Smith, C.; Chambers, B. An inventory of heavy metals inputs to agricultural soils in England and Wales. Sci. Total. Environ. 2003, 311, 205–219, doi:10.1016/s0048-9697(03)00139-6.
- François, F.; Lombard, C.; Guigner, J.-M.; Soreau, P.; Brian-Jaisson, F.; Martino, G.; Vandervennet, M.; Garcia, D.; Molinier, A.-L.; Pignol, D.; et al. Isolation and Characterization of Environmental Bacteria Capable of Extracellular Biosorption of Mercury. Appl. Environ. Microbiol. 2011, 78, 1097–1106, doi:10.1128/aem.06522-11.
- Samanta, A.; Bera, P.; Khatun, M.; Sinha, C.; Pal, P.; Lalee, A.; Mandal, A. An investigation on heavy metal tolerance and antibiotic resistance properties of bacterial strain Bacillus sp. isolated from municipal waste. J. Microbiol. Biotech. Res. 2012, 2, 178–189.
- Sobolev, D.; Begonia, M.F.T. Effects of Heavy Metal Contamination upon Soil Microbes: Lead-induced Changes in General and Denitrifying Microbial Communities as Evidenced by Molecular Markers. Int. J. Environ. Res. Public Heal. 2008, 5, 450–456, doi:10.3390/ijerph5050450.
- Hoostal, M.J.; Bidart-Bouzat, M.G.; Bouzat, J.L. Local adaptation of microbial communities to heavy metal stress in polluted sediments of Lake Erie. FEMS Microbiol. Ecol. 2008, 65, 156–168, doi:10.1111/j.1574-6941.2008.00522.x.
- Berg, J.; Brandt, K.K.; Abu Al-Soud, W.; Holm, P.E.; Hansen, L.H.; Sørensen, S.J.; Nybroe, O. Selection for Cu-Tolerant Bacterial Communities with Altered Composition, but Unaltered Richness, via Long-Term Cu Exposure. Appl. Environ. Microbiol. 2012, 78, 7438–7446, doi:10.1128/AEM.01071-12.
- Igiri, B.E.; Okoduwa, S.I.R.; Idoko, G.O.; Akabuogu, E.P.; Adeyi, A.O.; Ejiogu, I.K. Toxicity and Bioremediation of Heavy Metals Contaminated Ecosystem from Tannery Wastewater: A Review. J. Toxicol. 2018, 2018, 1–16, doi:10.1155/2018/2568038.
- Dickinson, A.; Power, A.; Hansen, M.; Brandt, K.; Piliposian, G.; Appleby, P.; O’Neill, P.; Jones, R.; Sierocinski, P.; Koskella, B.; et al. Heavy metal pollution and co-selection for antibiotic resistance: A microbial palaeontology approach. Environ. Int. 2019, 132, 105117, doi:10.1016/j.envint.2019.105117.
- Köhler, S.; Belkin, S.; Schmid, R.D. Reporter gene bioassays in environmental analysis. Anal. Bioanal. Chem. 2001, 366, 769–779, doi:10.1007/s002160051571.
- Tauriainen, S.; Virta, M.; Karp, M. Detecting bioavailable toxic metals and metalloids from natural water samples using luminescent sensor bacteria. Water Res. 2000, 34, 2661–2666, doi:10.1016/s0043-1354(00)00005-1.
- Flynn, H.; Meharg, A.; Bowyer, P.K.; Paton, G. Antimony bioavailability in mine soils. Environ. Pollut. 2003, 124, 93–100, doi:10.1016/s0269-7491(02)00411-6.
- Magrisso, S.; Erel, Y.; Belkin, S. Microbial reporters of metal bioavailability. Microb. Biotechnol. 2008, 1, 320–330, doi:10.1111/j.1751-7915.2008.00022.x.
- Rensing, C.; McDevitt, S.F. The copper metallome in prokaryotic cells. Met. Ions Life Sci. 2013, 12, 417–450.
- Meliani, A.; Bensoltane, A. Biofilm-Mediated Heavy Metals Bioremediation in PGPR Pseudomonas. J. Bioremediation Biodegrad. 2016, 7, doi:10.4172/2155-6199.1000370.
- Xu, Z.; Lei, Y.; Patel, J. Bioremediation of soluble heavy metals with recombinant Caulobacter crescentus. Bioeng. Bugs 2010, 1, 207–212, doi:10.4161/bbug.1.3.11246.
- Tabak, H.H.; Lens, P.N.L.; Van Hullebusch, E.D.; Dejonghe, W. Developments in Bioremediation of Soils and Sediments Polluted with Metals and Radionuclides–1. Microbial Processes and Mechanisms Affecting Bioremediation of Metal Contamination and Influencing Metal Toxicity and Transport. Rev. Environ. Sci. Biotechnol. 2005, 4, 115–156, doi:10.1007/s11157-005-2169-4.
- Akcil, A.; Erust, C.; Ozdemiroglu, S.; Fonti, V.; Beolchini, F. A review of approaches and techniques used in aquatic contaminated sediments: Metal removal and stabilization by chemical and biotechnological processes. J. Clean. Prod. 2015, 86, 24–36, doi:10.1016/j.jclepro.2014.08.009.
- Mergeay, M.; Van Houdt, R. Metal Response in Cupriavidus metallidurans Volume I: From Habitats to Genes and Proteins; Springer: Berlin, Germany, 2015.
- Mergeay, M.; Houba, C.; Gerits, J. Extrachromosomal Inheritance Controlling Resistance to Cobalt, Cadmium, Copper, and Zinc Ions: Evidence from Curing in a Pseudomonas. Arch. Int. Physiol. Biochim. 1978, 86, 440–441.
- Mergeay, M.; Nies, D.; Schlegel, H.G.; Gerits, J.; Charles, P.; Van Gijsegem, F. Alcaligenes eutrophus CH34 is a facultative chemolithotroph with plasmid-bound resistance to heavy metals. J. Bacteriol. 1985, 162, 328–334, doi:10.1128/jb.162.1.328-334.1985.
- Monsieurs, P.; Moors, H.; Van Houdt, R.; Janssen, P.J.; Janssen, A.; Coninx, I.; Mergeay, M.; Leys, N. Heavy metal resistance in Cupriavidus metallidurans CH34 is governed by an intricate transcriptional network. BioMetals 2011, 24, 1133–1151, doi:10.1007/s10534-011-9473-y.
- Nies, D.H. Microbial heavy-metal resistance. Appl. Microbiol. Biotechnol. 1999, 51, 730–750, doi:10.1007/s002530051457.
- Diels, L.; Van Roy, S.; Taghavi, S.; Van Houdt, R. From industrial sites to environmental applications with Cupriavidus metallidurans. Antonie Van Leeuwenhoek 2009, 96, 247–258, doi:10.1007/s10482-009-9361-4.
- Collard, J.M.; Corbisier, P.; Diels, L.; Dong, Q.; Jeanthon, C.; Mergeay, M.; Taghavi, S.; van der Lelie, D.; Wilmotte, A.; Wuertz, S. Plasmids for heavy metal resistance in Alcaligenes eutrophus CH34: Mechanisms and applications. FEMS Microbiol. Rev. 1994, 14, 405–414.
- Corbisier, P.; Thiry, E.; Diels, L. Bacterial Biosensors for the Toxicity Assessment of Solid Wastes. Environ Tox. Water Qual. Int. J. 1996, 11, 171–177.
- 27. Tibazarwa, C.; Corbisier, P.; Mench, M.; Bossus, A.; Solda, P.; Mergeay, M.; Wyns, L.; Van Der Lelie, D. A microbial biosensor to predict bioavailable nickel in soil and its transfer to plants. Environ. Pollut. 2001, 113, 19–26, doi:10.1016/s0269-7491(00)00177-9.
- Corbisier, P.; Van Der Lelie, D.; Borremans, B.; Provoost, A.; De Lorenzo, V.; Brown, N.L.; Lloyd, J.R.; Hobman, J.L.; Csöregi, E.; Johansson, G.; et al. Whole cell- and protein-based biosensors for the detection of bioavailable heavy metals in environmental samples. Anal. Chim. Acta 1999, 387, 235–244, doi:10.1016/s0003-2670(98)00725-9.
- Diels, L.; De Smet, M.; Hooyberghs, L.; Corbisier, P. Heavy Metals Bioremediation of Soil. Mol. Biotechnol. 1999, 12, 149–158, doi:10.1385/mb:12:2:149.
- Berezina, N.; Yada, B.; Lefebvre, R. From organic pollutants to bioplastics: Insights into the bioremediation of aromatic compounds by Cupriavidus necator. N. Biotechnol. 2015, 32, 47–53, doi:10.1016/j.nbt.2014.09.003.
- Janssen, P.J.; Van Houdt, R.; Moors, H.; Monsieurs, P.; Morin, N.; Michaux, A.; Benotmane, M.A.; Leys, N.; Vallaeys, T.; Lapidus, A.; et al. The Complete Genome Sequence of Cupriavidus metallidurans Strain CH34, a Master Survivalist in Harsh and Anthropogenic Environments. PLoS ONE 2010, 5, e10433, doi:10.1371/journal.pone.0010433.
- Lee, K.-Y.; Bosch, J.; Meckenstock, R.U. Use of metal-reducing bacteria for bioremediation of soil contaminated with mixed organic and inorganic pollutants. Environ. Geochem. Heal. 2011, 34, 135–142, doi:10.1007/s10653-011-9406-2.
- Köhler, C.; Lourenco, R.; Avelar, G.M.; Gomes, S.L. Extracytoplasmic function (ECF) sigma factor σF is involved in Caulobacter crescentus response to heavy metal stress. BMC Microbiol. 2012, 12, 210, doi:10.1186/1471-2180-12-210.
- Große, C.; Poehlein, A.; Blank, K.; Schwarzenberger, C.; Schleuder, G.; Herzberg, M.; Nies, D.H.; Grosse, C. The third pillar of metal homeostasis in Cupriavidus metallidurans CH34: Preferences are controlled by extracytoplasmic function sigma factors. Metallomics 2019, 11, 291–316, doi:10.1039/c8mt00299a.
- Osman, D.; Cavet, J.S. Copper Homeostasis in Bacteria. Adv. Appl. Microbiol. 2008, 65, 217–247.
- Argüello, J.M.; Raimunda, D.; Padilla-Benavides, T. Mechanisms of copper homeostasis in bacteria. Front. Microbiol. 2013, 3, 73, doi:10.3389/fcimb.2013.00073.
- Holmqvist, E.; Wagner, E.G.H. Impact of bacterial sRNAs in stress responses. Biochem. Soc. Trans. 2017, 45, 1203–1212, doi:10.1042/bst20160363.
- Hör, J.; Vogel, J. Global snapshots of bacterial RNA networks. EMBO J. 2016, 36, 245–247, doi:10.15252/embj.201696072.
- Chen, J.; Morita, T.; Gottesman, S. Regulation of Transcription Termination of Small RNAs and by Small RNAs: Molecular Mechanisms and Biological Functions. Front. Microbiol. 2019, 9, 201, doi:10.3389/fcimb.2019.00201.
- Dietrich, M.; Munke, R.; Gottschald, M.; Ziska, E.; Boettcher, J.P.; Mollenkopf, H.-J.; Friedrich, A. The effect of hfq on global gene expression and virulence in Neisseria gonorrhoeae. FEBS J. 2009, 276, 5507–5520, doi:10.1111/j.1742-4658.2009.07234.x.
- Sousa, S.A.; Ramos, C.G.; Moreira, L.M.; Leitão, J.H. The hfq gene is required for stress resistance and full virulence of Burkholderia cepacia to the nematode Caenorhabditis elegans. Microbiology 2009, 156, 896–908, doi:10.1099/mic.0.035139-0.
- Feliciano, J.R.; Grilo, A.M.; I Guerreiro, S.; Sousa, S.A.; Leitão, J.H. Hfq: A multifaceted RNA chaperone involved in virulence. Futur. Microbiol. 2016, 11, 137–151, doi:10.2217/fmb.15.128.
- Kakoschke, T.K.; Kakoschke, S.C.; Zeuzem, C.; Bouabe, H.; Adler, K.; Heesemann, J.; Rossier, O. The RNA Chaperone Hfq Is Essential for Virulence and Modulates the Expression of Four Adhesins in Yersinia enterocolitica. Sci. Rep. 2016, 6, 29275, doi:10.1038/srep29275.
- Fantappiè, L.; Metruccio, M.; Seib, K.L.; Oriente, F.; Cartocci, E.; Ferlicca, F.; Giuliani, M.M.; Scarlato, V.; Delany, I. The RNA Chaperone Hfq Is Involved in Stress Response and Virulence in Neisseria meningitidis and Is a Pleiotropic Regulator of Protein Expression. Infect. Immun. 2009, 77, 1842–1853, doi:10.1128/iai.01216-08.
- Wilson, J.W.; Ott, C.M.; Zu Bentrup, K.H.; Ramamurthy, R.; Quick, L.; Porwollik, S.; Cheng, P.; McClelland, M.; Tsaprailis, G.; Radabaugh, T.; et al. Space flight alters bacterial gene expression and virulence and reveals a role for global regulator Hfq. Proc. Natl. Acad. Sci. USA 2007, 104, 16299–16304, doi:10.1073/pnas.0707155104.
- Eshghi, A.; Becam, J.; Lambert, A.; Sismeiro, O.; Dillies, M.-A.; Jagla, B.; Wunder, E.A.J.; Ko, A.I.; Coppee, J.-Y.; Goarant, C.; et al. A Putative Regulatory Genetic Locus Modulates Virulence in the Pathogen Leptospira interrogans. Infect. Immun. 2014, 82, 2542–2552, doi:10.1128/iai.01803-14.
- Geng, J.; Song, Y.; Yang, L.; Feng, Y.; Qiu, Y.; Li, G.; Guo, J.; Bi, Y.; Qu, Y.; Wang, W.; et al. Involvement of the Post-Transcriptional Regulator Hfq in Yersinia pestis Virulence. PLoS ONE 2009, 4, e6213, doi:10.1371/journal.pone.0006213.
- Molina‐Santiago, C.; Daddaoua, A.; Gómez-Lozano, M.; Udaondo, Z.; Molin, S.; Ramos, J.L. Differential transcriptional response to antibiotics by Pseudomonas putida DOT-T1E. Environ. Microbiol. 2015, 17, 3251–3262, doi:10.1111/1462-2920.12775.
- Chen, Y.; Indurthi, D.C.; Jones, S.W.; Papoutsakis, E.T. Small RNAs in the Genus Clostridium. mBio 2011, 2, 00340–10, doi:10.1128/mBio.00340-10.
- Yu, J.; Schneiders, T. Tigecycline challenge triggers sRNA production in Salmonella enterica serovar Typhimurium. BMC Microbiol. 2012, 12, 195, doi:10.1186/1471-2180-12-195.
- Howden, B.P.; Beaume, M.; Harrison, P.F.; Hernandez, D.; Schrenzel, J.; Seemann, T.; Francois, P.; Stinear, T.P. Analysis of the Small RNA Transcriptional Response in Multidrug-Resistant Staphylococcus aureus after Antimicrobial Exposure. Antimicrob. Agents Chemother. 2013, 57, 3864–3874, doi:10.1128/aac.00263-13.
- Kim, K.-S.; Bak, G.; Lee, J. Systematic analysis of the role of bacterial Hfq-interacting sRNAs in the response to antibiotics. J. Antimicrob. Chemother. 2015, 70, 1659–68, doi:10.1093/jac/dkv042.
- Fröhlich, K.; Papenfort, K. Interplay of regulatory RNAs and mobile genetic elements in enteric pathogens. Mol. Microbiol. 2016, 101, 701–713, doi:10.1111/mmi.13428.
- Oglesby-Sherrouse, A.G.; Murphy, E.R. Iron-responsive bacterial small RNAs: Variations on a theme. Metallomics 2013, 5, 276–86, doi:10.1039/c3mt20224k.
- Chareyre, S.; Mandin, P. Bacterial Iron Homeostasis Regulation by sRNAs. Regul. RNA Bact. Archaea 2018, 6, 267–281, doi:10.1128/9781683670247.ch16.
- Sridhar, J.; Gayathri, M. Transcriptome based Identification of silver stress responsive sRNAs from Bacillus cereus ATCC14579. Bioinformation 2019, 15, 474–479, doi:10.6026/97320630015474.
- De Freitas, E.C.; Ucci, A.P.; Teixeira, E.C.; Pedroso, G.A.; Hilario, E.; Zocca, V.F.B.; De Paiva, G.B.; Ferreira, H.; Pedrolli, D.B.; Bertolini, M.C. The copper-inducible copAB operon in Xanthomonas citri subsp. citri is regulated at transcriptional and translational levels. Microbiology 2019, 165, 355–365, doi:10.1099/mic.0.000767.
- Lalaouna, D.; Baude, J.; Wu, Z.; Tomasini, A.; Chicher, J.; Marzi, S.; Vandenesch, F.; Romby, P.; Caldelari, I.; Moreau, K. RsaC sRNA modulates the oxidative stress response of Staphylococcus aureus during manganese starvation. Nucleic Acids Res. 2019, 47, 9871–9887, doi:10.1093/nar/gkz728.
- Chen, Y.; Xue, D.; Sun, W.; Han, J.; Li, J.; Gao, R.; Zhou, Z.; Zhang, W.; Chen, M.; Lin, M.; et al. sRNA OsiA Stabilizes Catalase mRNA during Oxidative Stress Response of Deincoccus radiodurans R1. Microorganisms 2019, 7, 422, doi:10.3390/microorganisms7100422.
- Peng, T.; Kan, J.; Lun, J.; Hu, Z. Identification of novel sRNAs involved in oxidative stress response in the fish pathogen Vibrio alginolyticus by transcriptome analysis. J. Fish Dis. 2018, 42, 277–291, doi:10.1111/jfd.12926.
- Olaya-Abril, A.; Luque-Almagro, V.M.; Pérez, M.D.; López, C.M.; Amil-Ruiz, F.; Cabello, P.; Sáez, L.P.; Moreno-Vivián, C.; Roldán, M.D. Putative small RNAs controlling detoxification of industrial cyanide-containing wastewaters by Pseudomonas pseudoalcaligenes CECT5344. PLoS ONE 2019, 14, e0212032, doi:10.1371/journal.pone.0212032.
- Innocenti, N.; Golumbeanu, M.; D’Herouel, A.F.; Lacoux, C.; Bonnin, R.A.; Kennedy, S.P.; Wessner, F.; Serror, P.; Bouloc, P.; Repoila, F.; et al. Whole-genome mapping of 5′ RNA ends in bacteria by tagged sequencing: A comprehensive view in Enterococcus faecalis. RNA 2015, 21, 1018–1030, doi:10.1261/rna.048470.114.
- Sharma, C.M.; Vogel, J. Differential RNA-seq: The approach behind and the biological insight gained. Curr. Opin. Microbiology 2014, 19, 97–105, doi:10.1016/j.mib.2014.06.010.
- Heidrich, N.; Bauriedl, S.; Barquist, L.; Li, L.; Schoen, C.; Vogel, J. The primary transcriptome of Neisseria meningitidis and its interaction with the RNA chaperone Hfq. Nucleic Acids Res. 2017, 45, 6147–6167, doi:10.1093/nar/gkx168.
- Vandecraen, J.; Monsieurs, P.; Mergeay, M.; Leys, N.; Aertsen, A.; Van Houdt, R. Zinc-Induced Transposition of Insertion Sequence Elements Contributes to Increased Adaptability of Cupriavidus metallidurans. Front. Microbiol. 2016, 7, 7, doi:10.3389/fmicb.2016.00359.
- Borremans, B.; Hobman, J.L.; Provoost, A.; Brown, N.L.; Van Der Lelie, D. Cloning and Functional Analysis of thepbr Lead Resistance Determinant of Ralstonia metallidurans CH34. J. Bacteriol. 2001, 183, 5651–5658, doi:10.1128/jb.183.19.5651-5658.2001.
- Große, C.; Grass, G.; Anton, A.; Franke, S.; Santos, A.N.; Lawley, B.; Brown, N.L.; Nies, D.H. Transcriptional Organization of the czcHeavy-Metal Homeostasis Determinant from Alcaligenes eutrophus. J. Bacteriol. 1999, 181, 2385–2393, doi:10.1128/jb.181.8.2385-2393.1999.
- Lomsadze, A.; Gemayel, K.; Tang, S.; Borodovsky, M. Modeling leaderless transcription and atypical genes results in more accurate gene prediction in prokaryotes. Genome Res. 2018, 28, 1079–1089, doi:10.1101/gr.230615.117.
- Schmidt, T.; Schlegel, H.G. Combined nickel-cobalt-cadmium resistance encoded by the ncc locus of Alcaligenes xylosoxidans 31A. J. Bacteriol. 1994, 176, 7045–7054, doi:10.1128/jb.176.22.7045-7054.1994.
- Hynninen, A.; Touzé, T.; Pitkänen, L.; Mengin-Lecreulx, D.; Virta, M.; Mengin-Lecreulx, M. An efflux transporter PbrA and a phosphatase PbrB cooperate in a lead-resistance mechanism in bacteria. Mol. Microbiol. 2009, 74, 384–394, doi:10.1111/j.1365-2958.2009.06868.x.
- Keshav, V.; Achilonu, I.; Dirr, H.W.; Kondiah, K. Recombinant expression and purification of a functional bacterial metallo-chaperone PbrD-fusion construct as a potential biosorbent for Pb(II). Protein Expr. Purif. 2019, 158, 27–35, doi:10.1016/j.pep.2019.02.008.
- Sass, A.M.; Van Acker, H.; Förstner, K.U.; Van Nieuwerburgh, F.; Deforce, D.; Vogel, J.; Coenye, T. Genome-wide transcription start site profiling in biofilm-grown Burkholderia cenocepacia J2315. BMC Genom. 2015, 16, 775, doi:10.1186/s12864-015-1993-3.
- Vockenhuber, M.-P.; Sharma, C.M.; Statt, M.G.; Schmidt, D.; Xu, Z.; Dietrich, S.; Liesegang, H.; Mathews, D.H.; Suess, B. Deep sequencing-based identification of small non-coding RNAs in Streptomyces coelicolor. RNA Boil. 2011, 8, 468–477, doi:10.4161/rna.8.3.14421.
- Kröger, C.; Dillon, S.C.; Cameron, A.D.S.; Papenfort, K.; Sivasankaran, S.K.; Hokamp, K.; Chao, Y.; Sittka, A.; Hébrard, M.; Haendler, K.; et al. The transcriptional landscape and small RNAs of Salmonella enterica serovar Typhimurium. Proc. Natl. Acad. Sci. USA 2012, 109, E1277–E1286, doi:10.1073/pnas.1201061109.
- Zhou, B.; Schrader, J.M.; Kalogeraki, V.S.; Abeliuk, E.; Dinh, C.B.; Pham, J.Q.; Cui, Z.Z.; Dill, D.L.; McAdams, H.H.; Shapiro, L. The Global Regulatory Architecture of Transcription during the Caulobacter Cell Cycle. PLoS Genet. 2015, 11, e1004831, doi:10.1371/journal.pgen.1004831.
- Schrader, J.M.; Zhou, B.; Li, G.-W.; Lasker, K.; Childers, W.S.; Williams, B.; Long, T.; Crosson, S.; McAdams, H.H.; Weissman, J.S.; et al. The Coding and Noncoding Architecture of the Caulobacter crescentus Genome. PLoS Genet. 2014, 10, e1004463, doi:10.1371/journal.pgen.1004463.
- Sharma, C.M.; Hoffmann, S.; Darfeuille, F.; Reignier, J.; Findeiß, S.; Sittka, A.; Chabas, S.; Reiche, K.; Hackermüller, J.; Reinhardt, R.; et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 2010, 464, 250–255, doi:10.1038/nature08756.
- Bischler, T.; Tan, H.S.; Nieselt, K.; Sharma, C.M. Differential RNA-seq (dRNA-seq) for annotation of transcriptional start sites and small RNAs in Helicobacter pylori. Methods 2015, 86, 89–101, doi:10.1016/j.ymeth.2015.06.012.
- Zheng, X.; Hu, G.; She, Z.-S.; Zhu, H. Leaderless genes in bacteria: Clue to the evolution of translation initiation mechanisms in prokaryotes. BMC Genom. 2011, 12, 361, doi:10.1186/1471-2164-12-361.
- Beck, H.J.; Moll, I. Leaderless mRNAs in the Spotlight: Ancient but Not Outdated! Microbiol. Spectr. 2018, 6, 155–170, doi:10.1128/9781683670247.ch10.
- Ao, W.; Gaudet, J.; Kent, W.J.; Muttumu, S.; Mango, S.E. Environmentally Induced Foregut Remodeling by PHA-4/FoxA and DAF-12/NHR. Science 2004, 305, 1743–1746.
- Li, J.; Qi, L.; Guo, Y.; Yue, L.; Li, Y.; Ge, W.; Wu, J.; Shi, W.; Dong, X. Global mapping transcriptional start sites revealed both transcriptional and post-transcriptional regulation of cold adaptation in the methanogenic archaeon Methanolobus psychrophilus. Sci. Rep. 2015, 5, 9209, doi:10.1038/srep09209.
- Mitschke, J.; Vioque, A.; Haas, F.B.; Hess, W.R.; Muro-Pastor, A.M. Dynamics of transcriptional start site selection during nitrogen stress-induced cell differentiation in Anabaena sp. PCC7120. Proc. Natl. Acad. Sci. USA 2011, 108, 20130–20135, doi:10.1073/pnas.1112724108.
- Thomason, M.K.; Bischler, T.; Eisenbart, S.K.; Förstner, K.U.; Zhang, A.; Herbig, A.; Nieselt, K.; Sharma, C.M.; Storz, G. Global Transcriptional Start Site Mapping Using Differential RNA Sequencing Reveals Novel Antisense RNAs in Escherichia coli. J. Bacteriol. 2014, 197, 18–28, doi:10.1128/jb.02096-14.
- Ko, J.-H.; Lee, S.-J.; Cho, B.; Lee, Y. Differential promoter usage ofinfAin response to cold shock in Escherichia coli. FEBS Lett. 2005, 580, 539–544, doi:10.1016/j.febslet.2005.12.066.
- Güell, M.; Yus, E.; Lluch-Senar, M.; Serrano, L. Bacterial transcriptomics: What is beyond the RNA horiz-ome? Nat. Rev. Genet. 2011, 9, 658–669, doi:10.1038/nrmicro2620.
- Lloréns-Rico, V.; Cano, J.; Kamminga, T.; Gil Garcia, R.; Latorre, A.; Chen, W.-H.; Bork, P.; Glass, J.I.; Serrano, L.; Lluch-Senar, M. Bacterial antisense RNAs are mainly the product of transcriptional noise. Sci. Adv. 2016, 2, e1501363, doi:10.1126/sciadv.1501363.
- Thomason, M.K.; Storz, G. Bacterial Antisense RNAs: How Many Are There, and What Are They Doing? Annu. Rev. Genet. 2010, 44, 167–188, doi:10.1146/annurev-genet-102209-163523.
- Eckweiler, D.; Häussler, S. Antisense transcription in Pseudomonas aeruginosa. Microbiology 2018, 164, 889–895, doi:10.1099/mic.0.000664.
- Nicolas, P.; Mäder, U.; Dervyn, E.; Rochat, T.; LeDuc, A.; Pigeonneau, N.; Bidnenko, E.; Marchadier, E.; Hoebeke, M.; Aymerich, S.; et al. Condition-Dependent Transcriptome Reveals High-Level Regulatory Architecture in Bacillus subtilis. Science 2012, 335, 1103–1106, doi:10.1126/science.1206848.
- Eiamphungporn, W.; Helmann, J.D. Extracytoplasmic function sigma factors regulate expression of the Bacillus subtilis yabE gene via a cis-acting antisense RNA. J. Bacteriol, 2009, 191, 1101–1105.
- Madjalani, N.; Cunning, C.; Sledjeski, D.; Elliott, T.; Gottesman, S. DsrA RNA regulates translation of RpoS message by an anti-antisense mechanism, independent of its action as an antisilencer of transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 12462–12467.
- Friedman, R.C.; Kalkhof, S.; Doppelt-Azeroual, O.; Mueller, S.A.; Chovancová, M.; Von Bergen, M.; Schwikowski, B. Common and phylogenetically widespread coding for peptides by bacterial small RNAs. BMC Genom. 2017, 18, 553, doi:10.1186/s12864-017-3932-y.
- Tsai, C.-H.; Liao, R.; Chou, B.; Palumbo, M.; Contreras, L.M. Genome-Wide Analyses in Bacteria Show Small-RNA Enrichment for Long and Conserved Intergenic Regions. J. Bacteriol. 2014, 197, 40–50, doi:10.1128/jb.02359-14.
- Sobrero, P.; Valverde, C. The bacterial protein Hfq: Much more than a mere RNA-binding factor. Crit. Rev. Microbiol. 2012, 38, 276–299, doi:10.3109/1040841x.2012.664540.

