 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rony Seger | + 2930 word(s) | 2930 | 2021-12-14 09:13:43 | | | |
| 2 | Lindsay Dong | + 1158 word(s) | 4088 | 2022-01-28 03:52:27 | | |
Video Upload Options
The mitogen-activated protein kinase (MAPK) cascades are key signaling components that transmit signals to many cellular processes. Each of the cascades operates by a sequential activation of protein kinases organized in three or more tiers that provide a seemingly linear signal transmission. However, the cellular effects regulated by each cascade may vary significantly. To achieve these diverse effects, the specificity of each cascade is extended by distinct regulators. Here we describe the importance of having distinct components in each tier of the cascades, particularly alternatively spliced isoforms of the MAPK components. This mode of regulation extends the cascade’s specificity and allows accurate, fine-tuned signaling outcomes that lead to proper cell fates.
1. Introduction
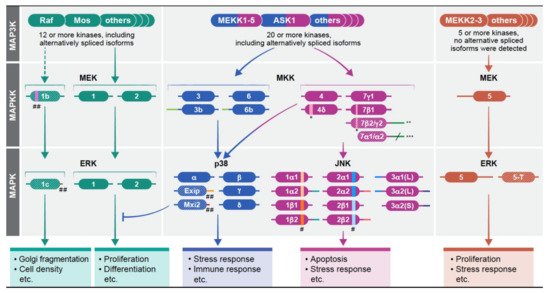
Mitogen-activated protein kinases (MAPKs) are a group of signaling proteins that regulate essentially all stimulated cellular processes [1][2]. Dysregulation of these kinases is involved in many diseases such as cancer and inflammation [3][4]. The MAPKs are operating within signaling cascades composed of three to five layers of protein kinases. MAP3K, MAPKK, and MAPK comprise the core and integral components of the cascade, whereas the upstream MAP4Ks and the downstream MAPKAPKs are added under some conditions. The signals of the cascade are transmitted via sequential phosphorylation and activation of the components in each layer. Four such cascades have been identified thus far: (i) Extracellular signal-regulated kinase 1/2 (ERK1/2) cascade that regulate mainly proliferation and differentiation [5][6][7][8]; (ii) cJun-N-terminal kinase (JNK) 1-3 (JNKs) cascade, that is mainly involved in stress response and apoptosis [9][10][11]; (iii) p38MAPKa-d (p38s) cascade, that regulates primarily stress response and the immune system [12][13][14]; and (iv) ERK5 that is involved in both proliferation and stress responses [15][16]. Even though the extracellular signals can be transmitted, at least to some extent, via all cascades, the strength and duration within each one of them are varied, dependent on the stimuli and cellular context. The MAPK signals use similar mechanisms of activation of these seemingly linear cascades, however, their output may be dramatically different between various stimuli. Therefore, it is clear that the specificity of the signals through the cascades must be tightly regulated in order to disseminate the proper outcomes. Indeed, many specificity-dictating processes regulate the cascades [17] including: (i) intensity and duration of the signals, which is regulated mainly by protein phosphatases [18]; (ii) scaffolding interactions which bring the components to close proximity with each other, and determine their localization [19]; (iii) crosstalk with other signaling pathways [20]; (iv) substrate-related regulation that involves distinct phosphorylation sites as well as substrates competition [21]; (v) dynamic subcellular localization of the MAPKs [22] and (vi) different effects of distinct components in each tier of the cascades (1). Here, we discuss the role of distinct gene products and alternatively spliced isoforms of the MAPKKs and MAPKs in determining signaling specificity of the cascades.
2. Multiple Isoforms Determine Signaling Specificity of the MAPK Cascades
The existence of various components with distinct function or regulation is an important way to extend the activities of the MAPK signaling cascades is through the existence of various components with distinct function or regulation within each tier of the cascades. This is mostly apparent in the MAP3K tier of the four cascades, in which 2-20 protein kinases may transmit distinct signals upon varying stimulations. Unlike the considerable number of components in the upper tiers, the number of components in the MAPKK and MAPK tiers is more limited, encoded by one to four genes that in some cases may have several protein products generated by alternative splicing. The activity and expression can be either similar to each other (e.g. JNK) or significantly different (e.g. ERK). In the case of different expression of the spliced forms, there is usually one main transcript, but other alternatively spiced variants exist and consist between 1-7% of the gene’s transcripts. In some cases, these minor components have distinct functions, and thereby may extend the specificity of the particular MAPK cascades. The gene products of the MAPKs are described in Figure 1 and Table 1 in Ref [23]). The signaling outcome of the different components in each tier of each cascade may also be regulated by their cognate expression levels. This point became apparent in the ERK components in knockout mice [24][25]. Whereas knockout of ERK1 has very little effect on mice survival [26], the knockout of ERK2 is embryonic lethal due to placental defects and defective mesoderm differentiation [27][28]. Similar expression-related changes were shown for other MAPKKs and MAPKs (e.g., MEK1/2, or p38α/β (24, 25), but not in the JNK cascade, in which the small differential effects are mainly due to distinct activities and regulation of the components [29][30]. It is important to mention that MEK5 and ERK5 do not have any notable alternatively spliced forms and their studies are very limited [31][32][33]. Therefore these isoforms will not be mentioned in the discussion below.
| Alternative Spliced Forms | Product of: | Sequence Changes | MW | Functional Changes | |
|---|---|---|---|---|---|
| MAPKKs | MEK1b | MAP2K1 (MEK1) | Deletion of 26 AA in subdomain 5 of MEK1. | 43.5 kDa | Reduced activity, change in substrate specificity. |
| MKK3b | MAP2K3 (MKK3) | Additional 29 amino acids N-terminal to MKK3. | 40 kDa | Slightly Elevates substrates’ phosphorylation. | |
| MKK6b | MAP2K6 (MKK6) | Additional 56 amino acids N terminal to MKK6. | 38.5 kDa | Affect substrate specificity. | |
| MKK7α1 | MAP2K7 (MKK7) | Deletion of the N terminal 89 AA of MKK7γ1. | 40 kDa | Reduced activity due to lack of JNK binding domain. | |
| MKK7α2 | MAP2K7 (MKK7) | Deletion of the N terminal 89 AA, + additional 33AA in the C terminus, compared to MKK7γ1. | 43.5 kDa | Reduced activity due to lack of JNK binding | |
| MKK7β1 | MAP2K7 (MKK7) | Deletion of 16 amino within the N terminus as compared to MKK7γ1. | 50 kDa | Distinct subcelluar localization, and lower binding to JNK due to lack of D domain. Distinct effect in cancers compared to MKK7γ1 | |
| MKK7β2 | MAP2K7 (MKK7) | Deletion of 16 amino within the N terminus + addition of 33 AA to the C terminus compared to MKK7γ1. | 53.5 kDa | Distinct subcelluar localization, a lower binding to JNK and distinct effect in cancers compared to MKK7γ1 | |
| MKK7γ2 | MAP2K7 (MKK7) | Addition of 33 AA to the C terminus, as compared to MKK7γ1. | 53.5 kDa | No known differences | |
| MAPK | ERK1c | MAPK1 (ERK1) | Change of the C terminal 40 AA with 18 other AA compared to ERK1. | 41.5 kDa | Reduced activity and change of substrate specificity as compared with ERK1. |
| Exip | MAPK14 (p38α) | Change of the C terminal 106 AA with 53 other AA compared to p38α. | 35.5 kDa | Reduced activity. Change in subcellular localization and protein interaction, as compared to p38α | |
| Mxi2 | MAPK14 (p38α) | Change of the C terminal 81 AA with 17 other AA compared to p38α. | 34 kDa | Reduced activity, change in substrate specificity, distinct protein interaction, and distinct regulation as compared to p38α. | |
| JNK1α1 | MAPK8 (JNK1) | Alternative exon 6a, Short C terminus | 46 kDa | Alternative substrate binding compared to β1 and β2. | |
| JNK1α2 | MAPK8 (JNK1) | Alternative exon 6a, long N terminus (43 AA) | 54 kDa | Alternative substrate binding compared to β1 and β2. | |
| JNK1β1 | MAPK8 (JNK1) | Alternative exon 6b, short C terminus | 46 kDa | Changes in expression level from α1 and α2. | |
| JNK1β2 | MAPK8 (JNK1) | Alternative exon 6b, long N terminus (43 AA) | 54 kDa | Changes in expression level from α1 and α2. | |
| JNK2α1 | MAPK9 (JNK2) | Alternative exon 6b, short C terminus | 46 kDa | Alternative substrate binding compared to β1 and β2. | |
| JNK2α2 | MAPK9 (JNK2) | Alternative exon 6b, long C terminus (42 AA) | 54 kDa | Alternative substrate binding compared to β1 and β2. | |
| JNK2β1 | MAPK9 (JNK2) | Alternative exon 6a, short C terminus | 46 kDa | Alternative substrate binding compared to α1 and α2. | |
| JNK2β2 | MAPK9 (JNK2) | Alternative exon 6a, long C terminus (42 AA | 54 kDa | Alternative substrate binding compared to α1 and α2. | |
| JNK3α1(L) | MAPK10 (JNK3) | Long N terminus (38 AA) short C terminus. | 50.5 kDa | Probable changes in protein interaction. | |
| JNK3α2(L) | MAPK10 (JNK3) | Long N terminus (38 AA) Long C terminus (42 AA). | 58.5 kDa | Probable changes in protein interaction. | |
| JNK3α2(S) | MAPK10 (JNK3) | Short N and long C termini (42 AA). | 54 kDa | Probable changes in protein interaction. | |
| ERK5-T | MAPK7 (ERK5) | Change of the C terminal 324 AA with 41 other AA. | 61 kDa | Distinct subcellular localization and protein binding compared to ERK5. |
3. Redundant and Unique Roles of MAPKKs of the JNK and p38 Cascades
The MAPKKs of the JNK cascade are MKK4 (that also phosphorylates p38s), and MKK7 that have two [34] and six [35] spliced isoforms respectively. They have one (MKK4) or two (MKK7) main isoforms, while the other are less abundant. The main isoforms are similar to each other, but have some distinct features that result in different activities. For example, MKK4 favors phosphorylation of the Tyr within the TPY domain of JNK, while MKK7 preferentially phosphorylates the Thr residue [36], indicating that the full activation of JNKs may sometimes require a combined activity of both kinases. In addition, MKK7 is specific to JNKs, but MKK4 can phosphorylate p38 as well [37]. Interestingly, a knockout of each of the two genes is embryonic lethal, supporting that they have unique functions [38][39][40]. Aside of the main isoforms, the other isoforms may provide additional regulation. While the main form, MKK4, prevents proliferation, MKK4d induces cell proliferation. The six isoforms of MKK7 [35] have distinct expression in different cells, and either MKK7g1 [41][42], or MKK7b1 [43]), are usually expressed more than the other, might serve as a main JNK activating kinase. The expression of MKK7b2 and g2 is lower, while MKK7a1/2 are lowly expressed. Besides their expression differences, MKK7a1/2 are also less active, and the distinct isoforms differ in their ability to bind scaffold proteins (e.g. Filamin [44] or JIP1 ([45]). Finally, during T cell activation, the MKK7 splicing is mediated by CELF2 favoring MKK7g1 isoform. This process is regulated by JNK, and thus induces a positive feedback upon stimulation [46].
The MAPKKs of the p38 cascade are MKK3, MKK6, and as mentioned above, occasionally also MKK4. Each one has two alternatively spliced proteins that are substantially expressed, but the expression level of the main isoforms is always higher than the expression of the others (MKK3b/6b), although vary between cell lines [47][48]. The human MKK3 and MKK6 are similar in their primary sequences, and in their ability to phosphorylate p38 [47], but seem to be activated by distinct stimuli, and preferentially (but not exclusively) phosphorylate distinct p38 isoforms [49][50][51]. In addition, it was shown that the balance between MKK3 and MKK6 mediates p38-associated resistance to cisplatin [52]. During development, MKK3/6 are mostly functionally redundant, as knockout of each of them individually does not affect viability, while simultaneous knockout of both is embryonic lethal due to problems in the hematopoietic system [53]. MKK3 and MKK6 also have cell type specific functions, such as in the differentiation of T cells [54], but those differences are not sufficient to induce embryonic lethality. No particular differences in activity have been demonstrated for the distinct alternatively spliced isoforms, and although the main isoforms are the most active in most cases, MKK3b and 6b may serve as the main p38 activators as well, phosphorylating all four p38 isoforms [55][56].
The MAPK components in the JNK cascades, JNK1 and JNK2, are each transcribed into four confirmed transcripts, and JNK3 has three [57][58]. The alternatively spliced isoforms of JNK1 and JNK2 at the protein level result from either the inclusion of alternative sequences in intron 6 (either α or β), or from different exons in the C-termini known as 1 or 2 [57][59]. The alternatively spliced isoforms of JNK3 have either elongated N or C termini ((JNK3α1(L), JNK3α2(L) or (JNK3α2(S)). The knockout of each of the genes alone results in viable animals with limited metabolic, immunological, and other pathologies [60][61]. Moreover, each one of the JNK proteins has similar catalytic activity towards most substrates, such as Ser34 of p53 [62]. This indicates that JNK1, JNK2, and to some extent JNK3 are mostly functionally redundant. However, the JNK1 and JNK2 are ubiquitously expressed, but their expression levels vary between tissues and cells. JNK3 is expressed almost exclusively in the brain and testis. The variable expression contributes to the limited differences in the single knockout animals, which results mostly from distinct signaling specificity [58][63][64]. Among several knockout outcomes, JNK1 knockout mice demonstrate a dysregulated neuronal differentiation during development [65]. Surprisingly, while JNK3 is mainly expressed in the brain, its knockout effects on the developing brain are more limited, showing mainly hippocampal neurogenesis malformations [66][67]. JNK2 does not seem to participate in these processes, but may play a role in carcinogenesis [68]. In addition, there are some cases in which the actions of JNK1 and JNK2 are cooperative or even synergistic, such as the development of skin keratinocytes [69] or UV- and arsenite-induced apoptosis [70]. The activities of the alternative spliced isoforms are usually similar, but it was shown that JNK2α3 has higher activity towards the transcription factor AP-1 than JNK2β3 [71]. Another study revealed that the short JNK proteins are less stable due to the interaction of the long isoforms with the scaffold protein JIP1 [72]. This stability modulation represents a new mechanism to regulate the JNK pathway. Thus, it is clear that the different JNK components can provide extended specificity to the cascade in response to various stimulations.
Four p38 MAPKs genes (a-d) have been identified in human, p38a has ~80% sequence similarity with p38b (also termed p38b2), while p38g has ~80% similarity with p38d, but the p38a/b pair has just 60% identity with the p38g/d pair [12][73]. The only splice variants reported are of p38a (Exip and Mxi2), which are both lowly expressed. All main gene-products can phosphorylate similar substrates and share similar regulation. Yet, they may display distinct properties and differ in their substrate specificity. Thus, it was shown that p38a/b phosphorylate MK2 and MK3 better than p38g/d [74], while the latter phosphorylate better the tau protein [75]. Whereas p38a is ubiquitously expressed, the expression of the products of the three other genes varies, and some are restricted to specific tissues. Knockout of p38a is embryonic lethal due to placental defects [76], while p38b-d knockout mice are viable and fertile [77][78]. The different effects may be mediated by distinct expression or by a minor difference in substrate repertoire. The specificity of the p38 is extended by the alternatively spliced isoforms of p38a. The first confirmed spliced isoform of p38a, was termed Exip (exon skip), that is considerably less abundant than the main p38a isoform [79][80]. Exip is not significantly phosphorylated on its activatory TGY motif due to lack of MAPKKs binding site, but can induce earlier apoptosis upon stress. The second lowly expressed spliced variant is Mxi2 (Max interactor), that affects c-Myc transcriptional activity [81]. It has a unique 17 amino acids in subdomain XI, and lacks the whole C-terminus [82]. Mxi2 is usually poorly expressed, but is found in relatively high amounts in mouse kidney, where its expression level is reduced by ischemia [83]. It was also shown that Mxi2 interacts with ERK1/2, regulating their non-stimulated nuclear translocation that leads to a sustained nuclear phosphorylation [84][85][86]. Interestingly, it was recently shown that Mxi2 is highly expressed in prostate cancer as well, where it increases the aggressiveness of the diseases, acting mainly by interacting with the Argonaut2/mir1285 complex that further regulate p53 activity [87].
4. The MEK1b-ERK1c axis
The main proteins translated by the ERK1 and ERK2 genes are the 44 and 42 kDa isoforms respectively. These isoforms usually share similar characteristics, although some differences between them do exist [88][89][90][91][92][93][94][95][96][97]. Aside of these main isoforms, initial studies reported the existence of additional, slightly different transcripts termed ERK1psi [98] and ERK2a [99], but these transcripts exist only on the RNA level. On the other hand, pan-ERK antibodies revealed an additional band of 46 kDa, tentatively termed ERK4 [98] in rat-derived cells. Later on, the band was found also in mice, but not in humans, and was further studied by our group. This latter study came as a consequence of in-gel kinase assay of rat cells, aiming to identify elevated kinase activity in transformed cells. One phosphorylated band at 46 kDa stood out as the most notable kinase activity that was even increased after Ras transformation [100]. This band was purified and cloned, giving rise to an alternatively spliced isoform of ERK1 with a 26 amino acid insertion (intron 7) inside the common docking motif (CD) of ERK1 of rat and mouse. The expression of this isoform was much lower (1-10%) than that of ERK1 and ERK2. Further characterization confirmed the similarity of this protein to the band originally identified as ERK4, and therefore was renamed as ERK1b. Under many circumstances, ERK1b behaves similarly to ERK1/2, but in Ras-transformed Rat1 cells, the expression level of ERK1b is elevated, and it is phosphorylated in a different kinetic than the main isoforms.
The lack of 46 kDa band in other organisms prompted a study aimed to identify whether any alternative spliced isoform is generated in humans. Using PCR, Aebersold et al demonstrated that inclusion of ERK1’s intron 7 does occur in human as well [101]. However, since in humans the intron contains an in-frame stop codon, the translation of this isoform resulted in a 42 kDa protein. This splicing event alters the C-terminal, containing 18 unique AA sequence just after the CD domain and not the rest of the C terminus of ERK1. Since this primate’s alternatively splice isoform was different from the rodent ERK1b, it was named ERK1c. This protein migrates to the same place as ERK2 on SDS-PAGE, and therefore cannot be detected by pan-ERK antibodies. However, the expression of this protein was confirmed using specific antibody to the unique ERK1c sequence, that detected low expression in various human cell [101]. Unlike the diffused subcellular localization of ERK1/2, ERK1c was found localized all over the cytoplasm in most cells, while in G2/M cells, it was localized primarily in the Golgi. In-depth study revealed that ERK1c regulates Golgi fragmentation, which was prevented by expression of dominant-negative ERK1c. Moreover, the expression and activity of ERK1c is increased in mitosis, following by its accumulation in the Golgi [102]. Indeed, small interfering RNA specific to ERK1c significantly attenuated, whereas ERK1c overexpression facilitated Golgi fragmentation. The main isoforms of ERK1 and ERK2 are active in different stages of mitosis, but do not significantly replace ERK1c -regulated part of the process .
One question that was unsolved at this stage was the way by which ERK1c is activated. Using the three MEK constructs, Shaul et al found that ERK1c is preferentially activated by MEK1b and not by MEK1 or MEK2 [103]. Interestingly, MEK1b was initially considered an inactive isoform, because it was unable to phosphorylate the main isoform of ERK1 and ERK2 [104]. Additionaly, Shaul et al showed that MEK1b phosphorylation and activity are preferentially stimulated by mitotic inducers (e.g. nocodazole, [105]) rather than growth factors or stress mediators (Figure 2). MEK1/2, on the other hand, preferentially target ERK1/2 and not ERK1c. Similar to ERK1c, MEK1b expression and activity are elevated during mitosis, though this isoform is localized primarily in the Golgi throughout the cell cycle. These results indicate that the ERK cascade can be divided into two routes: the classic MEK1/2-ERK1/2 that mediate most extracellular signals, and the splice-variants MEK1b-ERK1c route that specifically regulates mitotic Golgi fragmentation, and thus extends the specificity of the ERK cascade.
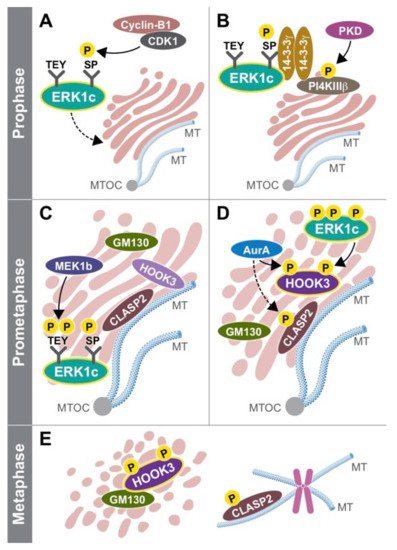
Since ERK1c is localized in the cytoplasm of cycling cells, Wortzel et al studied the molecular machinery that regulates this translocation. It was found that this translocation requires CDK1-induced phosphorylation of Ser343 of ERK1c specifically at prophase and prometaphase stages [106][107]. Then, phosphorylated ERK1c binds to a protein complex of PI4KIIIb and 14-3-3g that escort the kinase to the Golgi. The stability of this complex is regulated by protein kinase D (PKD)-mediated phosphorylation of PI4KIIIb. Thus, ERK1c joins other Golgi shuttling proteins at G2 to M phase to integrate Golgi- fragmentation regulators into one coherent pathway. In order to reveal the function of ERK1c, Wortzel et al recently studied what are the ERK1c-specific substrates that regulate Golgi fragmentation [108]. By screening of several putative Golgi proteins, HOOK3 was identified as a mediator of ERK1c-induced Golgi fragmentation (Figure 2 in Ref [23]). ERK1c phosphorylates HOOK3 on Ser238, which is a prerequisite for an additional phosphorylation on Ser707 by AuroraA kinase. In cycling cells, HOOK3 interacts with microtubules (MTs) and links them to the Golgi to induce the stabilization of the organelle. During mitosis, the double phosphorylation of HOOK3 by ERK1c and AuroraA allows the detachment of HOOK3 from the MTs, and its interaction with the Golgi architectural protein GM130. This switch in HOOK3’s interactions reduces the Golgi stability that permits Golgi fragmentation. Thus, ERK1c supports Golgi fragmentation process by shuttling into the Golgi towards mitosis, where it is activated by the resident MEK1b. The active ERK1c then phosphorylates several substrates including HOOK3 to set the stage for the fragmentation.
References
- Keshet, Y. and Seger, R. (2010) The MAP kinase signaling cascades: a system of hundreds of components regulates a diverse array of physiological functions. Methods Mol. Biol. 661, 3-38.
- Cargnello, M. and Roux, P.P. (2011) Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75, 50-83.
- Plotnikov, A., Zehorai, E., Procaccia, S. and Seger, R. (2011) The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 1813, 1619-1633.
- Lee, S., Rauch, J. and Kolch, W. (2020) Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 21, 1102.
- Maik-Rachline, G., Hacohen-Lev-Ran, A. and Seger, R. (2019) Nuclear ERK: Mechanism of Translocation, Substrates, and Role in Cancer. Int. J. Mol. Sci. 20, 1194.
- Lu, N. and Malemud, C.J. (2019) Extracellular Signal-Regulated Kinase: A Regulator of Cell Growth, Inflammation, Chondrocyte and Bone Cell Receptor-Mediated Gene Expression. Int. J. Mol. Sci. 20,
- Klomp, J.E., Klomp, J.A. and Der, C.J. (2021) The ERK mitogen-activated protein kinase signaling network: the final frontier in RAS signal transduction. Biochem. Soc. Trans. 49, 253-267.
- Lavoie, H., Gagnon, J. and Therrien, M. (2020) ERK signalling: a master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol. 21, 607-632.
- Hotamisligil, G.S. and Davis, R.J. (2016) Cell Signaling and Stress Responses. Cold Spring Harb. Perspect. Biol. 8,
- Ha, J., Kang, E., Seo, J. and Cho, S. (2019) Phosphorylation Dynamics of JNK Signaling: Effects of Dual-Specificity Phosphatases (DUSPs) on the JNK Pathway. Int. J. Mol. Sci. 20, 6157.
- Dhanasekaran, D.N. and Reddy, E.P. (2017) JNK-signaling: A multiplexing hub in programmed cell death. Genes Cancer 8, 682-694.
- Canovas, B. and Nebreda, A.R. (2021) Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol. 27, 1-21.
- Maik-Rachline, G., Lifshits, L. and Seger, R. (2020) Nuclear P38: Roles in Physiological and Pathological Processes and Regulation of Nuclear Translocation. Int. J. Mol. Sci. 21, 6102.
- Han, J., Wu, J. and Silke, J. (2020) An overview of mammalian p38 mitogen-activated protein kinases, central regulators of cell stress and receptor signaling. F1000Res 9,
- Hoang, V.T., Yan, T.J., Cavanaugh, J.E., Flaherty, P.T., Beckman, B.S. and Burow, M.E. (2017) Oncogenic signaling of MEK5-ERK5. Cancer Lett. 392, 51-59.
- Tubita, A., Lombardi, Z., Tusa, I., Dello Sbarba, P. and Rovida, E. (2020) Beyond Kinase Activity: ERK5 Nucleo-Cytoplasmic Shuttling as a Novel Target for Anticancer Therapy. Int. J. Mol. Sci. 21, 938.
- Shaul, Y.D. and Seger, R. (2007) The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta 1773, 1213-1226.
- Marshall, C.J. (1995) Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80, 179-185.
- Kolch, W. (2005) Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 6, 827-837.
- Nguyen, L.K., Kolch, W. and Kholodenko, B.N. (2013) When ubiquitination meets phosphorylation: a systems biology perspective of EGFR/MAPK signalling. Cell Commun Signal 11, 52.
- Kim, Y., Andreu, M.J., Lim, B., Chung, K., Terayama, M., Jimenez, G., Berg, C.A., Lu, H. and Shvartsman, S.Y. (2011) Gene regulation by MAPK substrate competition. Dev. Cell 20, 880-887.
- Flores, K. and Seger, R. (2013) Stimulated nuclear import by beta-like importins. F1000Prime Rep 5, 41.
- Maik-Rachline, G., Wortzel, I. and Seger, R. (2021) Alternative Splicing of MAPKs in the Regulation of Signaling Specificity. Cells 10, 3466.
- Chang, L. and Karin, M. (2001) Mammalian MAP kinase signalling cascades. Nature 410, 37-40.
- Kuida, K. and Boucher, D.M. (2004) Functions of MAP kinases: insights from gene-targeting studies. J Biochem (Tokyo) 135, 653-656.
- Pages, G., Guerin, S., Grall, D., Bonino, F., Smith, A., Anjuere, F., Auberger, P. and Pouyssegur, J. (1999) Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science 286, 1374-1377.
- Yao, Y., Li, W., Wu, J., Germann, U.A., Su, M.S., Kuida, K. and Boucher, D.M. (2003) Extracellular signal-regulated kinase 2 is necessary for mesoderm differentiation. Proc. Natl. Acad. Sci. U. S. A. 100, 12759-12764.
- Hatano, N., Mori, Y., Oh-hora, M., Kosugi, A., Fujikawa, T., Nakai, N., Niwa, H., Miyazaki, J., Hamaoka, T. and Ogata, M. (2003) Essential role for ERK2 mitogen-activated protein kinase in placental development. Genes Cells 8, 847-856.
- Dong, C., Yang, D.D., Wysk, M., Whitmarsh, A.J., Davis, R.J. and Flavell, R.A. (1998) Defective T cell differentiation in the absence of Jnk1. Science 282, 2092-2095.
- Yang, D.D., Conze, D., Whitmarsh, A.J., Barrett, T., Davis, R.J., Rincon, M. and Flavell, R.A. (1998) Differentiation of CD4+ T cells to Th1 cells requires MAP kinase JNK2. Immunity 9, 575-585.
- Cameron, S.J., Abe, J., Malik, S., Che, W. and Yang, J. (2004) Differential role of MEK5alpha and MEK5beta in BMK1/ERK5 activation. J. Biol. Chem. 279, 1506-1512.
- English, J.M., Vanderbilt, C.A., Xu, S., Marcus, S. and Cobb, M.H. (1995) Isolation of MEK5 and differential expression of alternatively spliced forms. J. Biol. Chem. 270, 28897-28902.
- Nishimoto, S. and Nishida, E. (2006) MAPK signalling: ERK5 versus ERK1/2. EMBO Rep 7, 782-786.
- Haeusgen, W., Tueffers, L., Herdegen, T. and Waetzig, V. (2014) Map2k4delta - identification and functional characterization of a novel Map2k4 splice variant. Biochim. Biophys. Acta 1843, 875-884.
- Tournier, C., Whitmarsh, A.J., Cavanagh, J., Barrett, T. and Davis, R.J. (1999) The MKK7 gene encodes a group of c-Jun NH2-terminal kinase kinases. Mol. Cell. Biol. 19, 1569-1581.
- Lawler, S., Fleming, Y., Goedert, M. and Cohen, P. (1998) Synergistic activation of SAPK1/JNK1 by two MAP kinase kinases in vitro. Curr. Biol. 8, 1387-1390.
- Derijard, B., Raingeaud, J., Barrett, T., Wu, I.H., Han, J., Ulevitch, R.J. and Davis, R.J. (1995) Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms [published erratum appears in Science 1995 Jul 7;269(5220):17]. Science 267, 682-685.
- Ganiatsas, S., Kwee, L., Fujiwara, Y., Perkins, A., Ikeda, T., Labow, M.A. and Zon, L.I. (1998) SEK1 deficiency reveals mitogen-activated protein kinase cascade crossregulation and leads to abnormal hepatogenesis. Proc. Natl. Acad. Sci. U. S. A. 95, 6881-6886.
- Haeusgen, W., Herdegen, T. and Waetzig, V. (2011) The bottleneck of JNK signaling: molecular and functional characteristics of MKK4 and MKK7. Eur. J. Cell Biol. 90, 536-544.
- Asaoka, Y. and Nishina, H. (2010) Diverse physiological functions of MKK4 and MKK7 during early embryogenesis. J. Biochem. 148, 393-401.
- Haeusgen, W., Herdegen, T. and Waetzig, V. (2011) MKK7gamma1 reverses nerve growth factor signals: proliferation and cell death instead of neuritogenesis and protection. Cell. Signal. 23, 1281-1290.
- Gibson, E.S., Woolfrey, K.M., Li, H., Hogan, P.G., Nemenoff, R.A., Heasley, L.E. and Dell'Acqua, M.L. (2019) Subcellular Localization and Activity of the Mitogen-Activated Protein Kinase Kinase 7 (MKK7) gamma Isoform are Regulated through Binding to the Phosphatase Calcineurin. Mol. Pharmacol. 95, 20-32.
- Ray, D., Yun, Y.C., Idris, M., Cheng, S., Boot, A., Iain, T.B.H., Rozen, S.G., Tan, P. and Epstein, D.M. (2020) A tumor-associated splice-isoform of MAP2K7 drives dedifferentiation in MBNL1-low cancers via JNK activation. Proc. Natl. Acad. Sci. U. S. A. 117, 16391-16400.
- Nakagawa, K., Sugahara, M., Yamasaki, T., Kajiho, H., Takahashi, S., Hirayama, J., Minami, Y., Ohta, Y., Watanabe, T., Hata, Y., Katada, T. and Nishina, H. (2010) Filamin associates with stress signalling kinases MKK7 and MKK4 and regulates JNK activation. Biochem. J. 427, 237-245.
- Haeusgen, W., Boehm, R., Zhao, Y., Herdegen, T. and Waetzig, V. (2009) Specific activities of individual c-Jun N-terminal kinases in the brain. Neuroscience 161, 951-959.
- Martinez, N.M., Agosto, L., Qiu, J., Mallory, M.J., Gazzara, M.R., Barash, Y., Fu, X.D. and Lynch, K.W. (2015) Widespread JNK-dependent alternative splicing induces a positive feedback loop through CELF2-mediated regulation of MKK7 during T-cell activation. Genes Dev. 29, 2054-2066.
- Han, J., Lee, J.D., Jiang, Y., Li, Z., Feng, L. and Ulevitch, R.J. (1996) Characterization of the structure and function of a novel MAP kinase kinase (MKK6). J. Biol. Chem. 271, 2886-2891.
- Moriguchi, T., Toyoshima, F., Gotoh, Y., Iwamatsu, A., Irie, K., Mori, E., Kuroyanagi, N., Hagiwara, M., Matsumoto, K. and Nishida, E. (1996) Purification and identification of a major activator for p38 from osmotically shocked cells. Activation of mitogen-activated protein kinase kinase 6 by osmotic shock, tumor necrosis factor-alpha, and H2O2. J. Biol. Chem. 271, 26981-26988.
- Enslen, H., Raingeaud, J. and Davis, R.J. (1998) Selective activation of p38 mitogen-activated protein (MAP) kinase isoforms by the MAP kinase kinases MKK3 and MKK6. J. Biol. Chem. 273, 1741-1748.
- Keesler, G.A., Bray, J., Hunt, J., Johnson, D.A., Gleason, T., Yao, Z., Wang, S.W., Parker, C., Yamane, H., Cole, C. and Lichenstein, H.S. (1998) Purification and activation of recombinant p38 isoforms alpha, beta, gamma, and delta. Protein Expr. Purif. 14, 221-228.
- Remy, G., Risco, A.M., Inesta-Vaquera, F.A., Gonzalez-Teran, B., Sabio, G., Davis, R.J. and Cuenda, A. (2010) Differential activation of p38MAPK isoforms by MKK6 and MKK3. Cell. Signal. 22, 660-667.
- Galan-Moya, E.M., de la Cruz-Morcillo, M.A., Llanos Valero, M., Callejas-Valera, J.L., Melgar-Rojas, P., Hernadez Losa, J., Salcedo, M., Fernandez-Aramburo, A., Ramon y Cajal, S. and Sanchez-Prieto, R. (2011) Balance between MKK6 and MKK3 mediates p38 MAPK associated resistance to cisplatin in NSCLC. PLoS One 6, e28406.
- Brancho, D., Tanaka, N., Jaeschke, A., Ventura, J.J., Kelkar, N., Tanaka, Y., Kyuuma, M., Takeshita, T., Flavell, R.A. and Davis, R.J. (2003) Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 17, 1969-1978.
- Tanaka, N., Kamanaka, M., Enslen, H., Dong, C., Wysk, M., Davis, R.J. and Flavell, R.A. (2002) Differential involvement of p38 mitogen-activated protein kinase kinases MKK3 and MKK6 in T-cell apoptosis. EMBO Rep 3, 785-791.
- Huang, S., Jiang, Y., Li, Z., Nishida, E., Mathias, P., Lin, S., Ulevitch, R.J., Nemerow, G.R. and Han, J. (1997) Apoptosis signaling pathway in T cells is composed of ICE/Ced-3 family proteases and MAP kinase kinase 6b. Immunity 6, 739-749.
- Chen, Y., Shi, G., Xia, W., Kong, C., Zhao, S., Gaw, A.F., Chen, E.Y., Yang, G.P., Giaccia, A.J., Le, Q.T. and Koong, A.C. (2004) Identification of hypoxia-regulated proteins in head and neck cancer by proteomic and tissue array profiling. Cancer Res. 64, 7302-7310.
- Zeke, A., Misheva, M., Remenyi, A. and Bogoyevitch, M.A. (2016) JNK Signaling: Regulation and Functions Based on Complex Protein-Protein Partnerships. Microbiol. Mol. Biol. Rev. 80, 793-835.
- Gupta, S., Barrett, T., Whitmarsh, A.J., Cavanagh, J., Sluss, H.K., Derijard, B. and Davis, R.J. (1996) Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 15, 2760-2770.
- Barr, R.K. and Bogoyevitch, M.A. (2001) The c-Jun N-terminal protein kinase family of mitogen-activated protein kinases (JNK MAPKs). Int. J. Biochem. Cell Biol. 33, 1047-1063.
- Sabapathy, K., Jochum, W., Hochedlinger, K., Chang, L., Karin, M. and Wagner, E.F. (1999) Defective neural tube morphogenesis and altered apoptosis in the absence of both JNK1 and JNK2. Mech. Dev. 89, 115-124.
- Kuan, C.Y., Yang, D.D., Samanta Roy, D.R., Davis, R.J., Rakic, P. and Flavell, R.A. (1999) The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron 22, 667-676.
- Hu, M.C., Qiu, W.R. and Wang, Y.P. (1997) JNK1, JNK2 and JNK3 are p53 N-terminal serine 34 kinases. Oncogene 15, 2277-2287.
- Liu, J., Minemoto, Y. and Lin, A. (2004) c-Jun N-terminal protein kinase 1 (JNK1), but not JNK2, is essential for tumor necrosis factor alpha-induced c-Jun kinase activation and apoptosis. Mol. Cell. Biol. 24, 10844-10856.
- Sabapathy, K., Hochedlinger, K., Nam, S.Y., Bauer, A., Karin, M. and Wagner, E.F. (2004) Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun-dependent cell proliferation. Mol. Cell 15, 713-725.
- Amura, C.R., Marek, L., Winn, R.A. and Heasley, L.E. (2005) Inhibited neurogenesis in JNK1-deficient embryonic stem cells. Mol. Cell. Biol. 25, 10791-10802.
- Castro-Torres, R.D., Landa, J., Rabaza, M., Busquets, O., Olloquequi, J., Ettcheto, M., Beas-Zarate, C., Folch, J., Camins, A., Auladell, C. and Verdaguer, E. (2019) JNK Isoforms Are Involved in the Control of Adult Hippocampal Neurogenesis in Mice, Both in Physiological Conditions and in an Experimental Model of Temporal Lobe Epilepsy. Mol. Neurobiol. 56, 5856-5865.
- Nakano, R., Nakayama, T. and Sugiya, H. (2020) Biological Properties of JNK3 and Its Function in Neurons, Astrocytes, Pancreatic beta-Cells and Cardiovascular Cells. Cells 9,
- Nielsen, C., Thastrup, J., Bottzauw, T., Jaattela, M. and Kallunki, T. (2007) c-Jun NH2-terminal kinase 2 is required for Ras transformation independently of activator protein 1. Cancer Res. 67, 178-185.
- Schumacher, M., et al. (2014) Efficient keratinocyte differentiation strictly depends on JNK-induced soluble factors in fibroblasts. J. Invest. Dermatol. 134, 1332-1341.
- Zhang, D., Song, L., Li, J., Wu, K. and Huang, C. (2006) Coordination of JNK1 and JNK2 is critical for GADD45alpha induction and its mediated cell apoptosis in arsenite responses. J. Biol. Chem. 281, 34113-34123.
- Wang, P., Xiong, Y., Ma, C., Shi, T. and Ma, D. (2010) Molecular cloning and characterization of novel human JNK2 (MAPK9) transcript variants that show different stimulation activities on AP-1. BMB Rep 43, 738-743.
- Yang, J.Y., Moulin, N., van Bemmelen, M.X., Dubuis, G., Tawadros, T., Haefliger, J.A., Waeber, G. and Widmann, C. (2007) Splice variant-specific stabilization of JNKs by IB1/JIP1. Cell. Signal. 19, 2201-2207.
- Cuenda, A. and Rousseau, S. (2007) p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 1773, 1358-1375.
- Goedert, M., Cuenda, A., Craxton, M., Jakes, R. and Cohen, P. (1997) Activation of the novel stress-activated protein kinase SAPK4 by cytokines and cellular stresses is mediated by SKK3 (MKK6); comparison of its substrate specificity with that of other SAP kinases. EMBO J. 16, 3563-3571.
- Goedert, M., Hasegawa, M., Jakes, R., Lawler, S., Cuenda, A. and Cohen, P. (1997) Phosphorylation of microtubule-associated protein tau by stress-activated protein kinases. FEBS Lett. 409, 57-62.
- Gupta, J. and Nebreda, A.R. (2015) Roles of p38alpha mitogen-activated protein kinase in mouse models of inflammatory diseases and cancer. FEBS J 282, 1841-1857.
- Beardmore, V.A., Hinton, H.J., Eftychi, C., Apostolaki, M., Armaka, M., Darragh, J., McIlrath, J., Carr, J.M., Armit, L.J., Clacher, C., Malone, L., Kollias, G. and Arthur, J.S. (2005) Generation and characterization of p38beta (MAPK11) gene-targeted mice. Mol. Cell. Biol. 25, 10454-10464.
- Sabio, G., Arthur, J.S., Kuma, Y., Peggie, M., Carr, J., Murray-Tait, V., Centeno, F., Goedert, M., Morrice, N.A. and Cuenda, A. (2005) p38gamma regulates the localisation of SAP97 in the cytoskeleton by modulating its interaction with GKAP. EMBO J. 24, 1134-1145.
- Sudo, T., Yagasaki, Y., Hama, H., Watanabe, N. and Osada, H. (2002) Exip, a new alternative splicing variant of p38 alpha, can induce an earlier onset of apoptosis in HeLa cells. Biochem. Biophys. Res. Commun. 291, 838-843.
- Yagasaki, Y., Sudo, T. and Osada, H. (2004) Exip, a splicing variant of p38alpha, participates in interleukin-1 receptor proximal complex and downregulates NF-kappaB pathway. FEBS Lett. 575, 136-140.
- Zervos, A.S., Faccio, L., Gatto, J.P., Kyriakis, J.M. and Brent, R. (1995) Mxi2, a mitogen-activated protein kinase that recognizes and phosphorylates Max protein. Proc. Natl. Acad. Sci. U. S. A. 92, 10531-10534.
- Sanz, V., Arozarena, I. and Crespo, P. (2000) Distinct carboxy-termini confer divergent characteristics to the mitogen-activated protein kinase p38alpha and its splice isoform Mxi2. FEBS Lett. 474, 169-174.
- Faccio, L., Chen, A., Fusco, C., Martinotti, S., Bonventre, J.V. and Zervos, A.S. (2000) Mxi2, a splice variant of p38 stress-activated kinase, is a distal nephron protein regulated with kidney ischemia. Am. J. Physiol. Cell Physiol. 278, C781-790.
- Casar, B., Sanz-Moreno, V., Yazicioglu, M.N., Rodriguez, J., Berciano, M.T., Lafarga, M., Cobb, M.H. and Crespo, P. (2007) Mxi2 promotes stimulus-independent ERK nuclear translocation. EMBO J. 26, 635-646.
- Sanz-Moreno, V., Casar, B. and Crespo, P. (2003) p38alpha isoform Mxi2 binds to extracellular signal-regulated kinase 1 and 2 mitogen-activated protein kinase and regulates its nuclear activity by sustaining its phosphorylation levels. Mol. Cell. Biol. 23, 3079-3090.
- Casar, B., Rodriguez, J., Gibor, G., Seger, R. and Crespo, P. (2012) Mxi2 sustains ERK1/2 phosphorylation in the nucleus by preventing ERK1/2 binding to phosphatases. Biochem. J. 441, 571-578.
- KÖditz, B., Fries, J.W.U., GÖbel, H., Paffenholz, P., Richter, K., Heidenreich, A. and Von Brandenstein, M. (2020) Mxi-2 Dependent Regulation of p53 in Prostate Cancer. Anticancer Res. 40, 5539-5544.
- O'Brien, D.E., Alter, B.J., Satomoto, M., Morgan, C.D., Davidson, S., Vogt, S.K., Norman, M.E., Gereau, G.B., Demaro, J.A., 3rd, Landreth, G.E., Golden, J.P. and Gereau, R.W.t. (2015) ERK2 alone drives inflammatory pain but cooperates with ERK1 in sensory neuron survival. J. Neurosci. 35, 9491-9507.
- Bar-Gill, A.B., Efergan, A., Seger, R., Fukuda, M. and Sagi-Eisenberg, R. (2013) The extra-cellular signal regulated kinases ERK1 and ERK2 segregate displaying distinct spatiotemporal characteristics in activated mast cells. Biochim. Biophys. Acta 1833, 2070-2082.
- Guegan, J.P., Ezan, F., Theret, N., Langouet, S. and Baffet, G. (2013) MAPK signaling in cisplatin-induced death: predominant role of ERK1 over ERK2 in human hepatocellular carcinoma cells. Carcinogenesis 34, 38-47.
- von Thun, A., Birtwistle, M., Kalna, G., Grindlay, J., Strachan, D., Kolch, W., von Kriegsheim, A. and Norman, J.C. (2012) ERK2 drives tumour cell migration in three-dimensional microenvironments by suppressing expression of Rab17 and liprin-beta2. J. Cell Sci. 125, 1465-1477.
- Shin, S., Dimitri, C.A., Yoon, S.O., Dowdle, W. and Blenis, J. (2010) ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Mol. Cell 38, 114-127.
- Liu, X., Yan, S., Zhou, T., Terada, Y. and Erikson, R.L. (2004) The MAP kinase pathway is required for entry into mitosis and cell survival. Oncogene 23, 763-776.
- Yu, S., Zhang, M., Huang, L., Ma, Z., Gong, X., Liu, W., Zhang, J., Chen, L., Yu, Z., Zhao, W. and Liu, Y. (2019) ERK1 indicates good prognosis and inhibits breast cancer progression by suppressing YAP1 signaling. Aging (Albany NY) 11, 12295-12314.
- Crowe, M.S., Zavorotinskaya, T., Voliva, C.F., Shirley, M.D., Wang, Y., Ruddy, D.A., Rakiec, D.P., Engelman, J.A., Stuart, D.D. and Freeman, A.K. (2021) RAF-mutant melanomas differentially depend on ERK2 over ERK1 to support aberrant MAPK pathway activation and cell proliferation. Mol. Cancer Res.
- Marchi, M., D'Antoni, A., Formentini, I., Parra, R., Brambilla, R., Ratto, G.M. and Costa, M. (2008) The N-terminal domain of ERK1 accounts for the functional differences with ERK2. PLoS One 3, e3873.
- Crowe, M.S., Zavorotinskaya, T., Voliva, C.F., Shirley, M.D., Wang, Y., Ruddy, D.A., Rakiec, D.P., Engelman, J.A., Stuart, D.D. and Freeman, A.K. (2021) RAF-Mutant Melanomas Differentially Depend on ERK2 Over ERK1 to Support Aberrant MAPK Pathway Activation and Cell Proliferation. Mol. Cancer Res. 19, 1063-1075.
- Boulton, T.G., Nye, S.H., Robbins, D.J., Ip, N.Y., Radziejewska, E., Morgenbesser, S.D., DePinho, R.A., Panayotatos, N., Cobb, M.H. and Yancopoulos, G.D. (1991) ERK's: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell 65, 663-675.
- Gonzalez, F.A., Raden, D.L., Rigby, M.R. and Davis, R.J. (1992) Heterogeneous expression of four MAP kinase isoforms in human tissues. FEBS Lett. 304, 170-178.
- Yung, Y., Yao, Z., Hanoch, T. and Seger, R. (2000) ERK1b, a 46-kDa ERK Isoform That Is Differentially Regulated by MEK. Cell Biol. Int. Accepted,
- Aebersold, D.M., Shaul, Y.D., Yung, Y., Yarom, N., Yao, Z., Hanoch, T. and Seger, R. (2004) Extracellular signal-regulated kinase 1c (ERK1c), a novel 42-kilodalton ERK, demonstrates unique modes of regulation, localization, and function. Mol. Cell. Biol. 24, 10000-10015.
- Shaul, Y.D. and Seger, R. (2006) ERK1c regulates Golgi fragmentation during mitosis. J. Cell Biol. 172, 885-897.
- Shaul, Y.D., Gibor, G., Plotnikov, A. and Seger, R. (2009) Specific phosphorylation and activation of ERK1c by MEK1b: a unique route in the ERK cascade. Genes Dev. 23, 1779-1790.
- Zheng, C.F. and Guan, K.L. (1993) Properties of MEKs, the kinases that phosphorylate and activate the extracellular signal-regulated kinases. J. Biol. Chem. 268, 23933-23939.
- Seger, R. and Krebs, E.G. (1995) The MAPK signaling cascade. FASEB J. 9, 726-735.
- Wortzel, I., Hanoch, T., Porat, Z., Hausser, A. and Seger, R. (2015) Mitotic Golgi translocation of ERK1c is mediated by a PI4KIIIbeta-14-3-3gamma shuttling complex. J. Cell Sci. 128, 4083-4095.
- Wortzel, I., Koifman, G., Rotter, V., Seger, R. and Porat, Z. (2017) High Throughput Analysis of Golgi Structure by Imaging Flow Cytometry. Sci. Rep. 7, 788.
- Wortzel, I., Maik-Rachline, G., Yadav, S.S., Hanoch, T. and Seger, R. (2021) Mitotic HOOK3 phosphorylation by ERK1c drives microtubule-dependent Golgi destabilization and fragmentation. iScience 24, 102670.

