 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hamid Al-Jamal | + 2417 word(s) | 2417 | 2022-01-27 04:03:34 | | | |
| 2 | Vicky Zhou | Meta information modification | 2417 | 2022-01-27 13:10:13 | | | | |
| 3 | Vicky Zhou | + 1 word(s) | 2418 | 2022-01-28 06:18:32 | | | | |
| 4 | Vicky Zhou | + 22 word(s) | 2439 | 2022-01-28 06:24:24 | | |
Video Upload Options
Iron homeostasis is regulated by hepcidin, a hepatic hormone that controls dietary iron absorption and plasma iron concentration. Hepcidin binds to the only known iron export protein, ferroportin (FPN), which regulates its expression. The major factors that implicate hepcidin regulation include iron stores, hypoxia, inflammation, and erythropoiesis. When erythropoietic activity is suppressed, hepcidin expression is hampered, leading to deficiency, thus causing an iron overload in iron-loading anemia, such as β-thalassemia. Iron overload is the principal cause of mortality and morbidity in β-thalassemia patients with or without blood transfusion dependence. In the case of thalassemia major, the primary cause of iron overload is blood transfusion. In contrast, iron overload is attributed to hepcidin deficiency and hyperabsorption of dietary iron in non-transfusion thalassemia. Beta-thalassemia patients showed marked hepcidin suppression, anemia, iron overload, and ineffective erythropoiesis (IE). Recent molecular research has prompted the discovery of new diagnostic markers and therapeutic targets for several diseases, including β-thalassemia.
1. Thalassemia Syndrome
2. Beta-Thalassemia
2. Hepcidin Expression in β-Thalassemia
2.1. Hepcidin Regulation in β-Thalassemia
2.2. Regulatory Effect of Hepcidin Transcription
2.3. Hepcidin Therapeutics in β-Thalassemia
3. TGF-β/SMAD Signaling
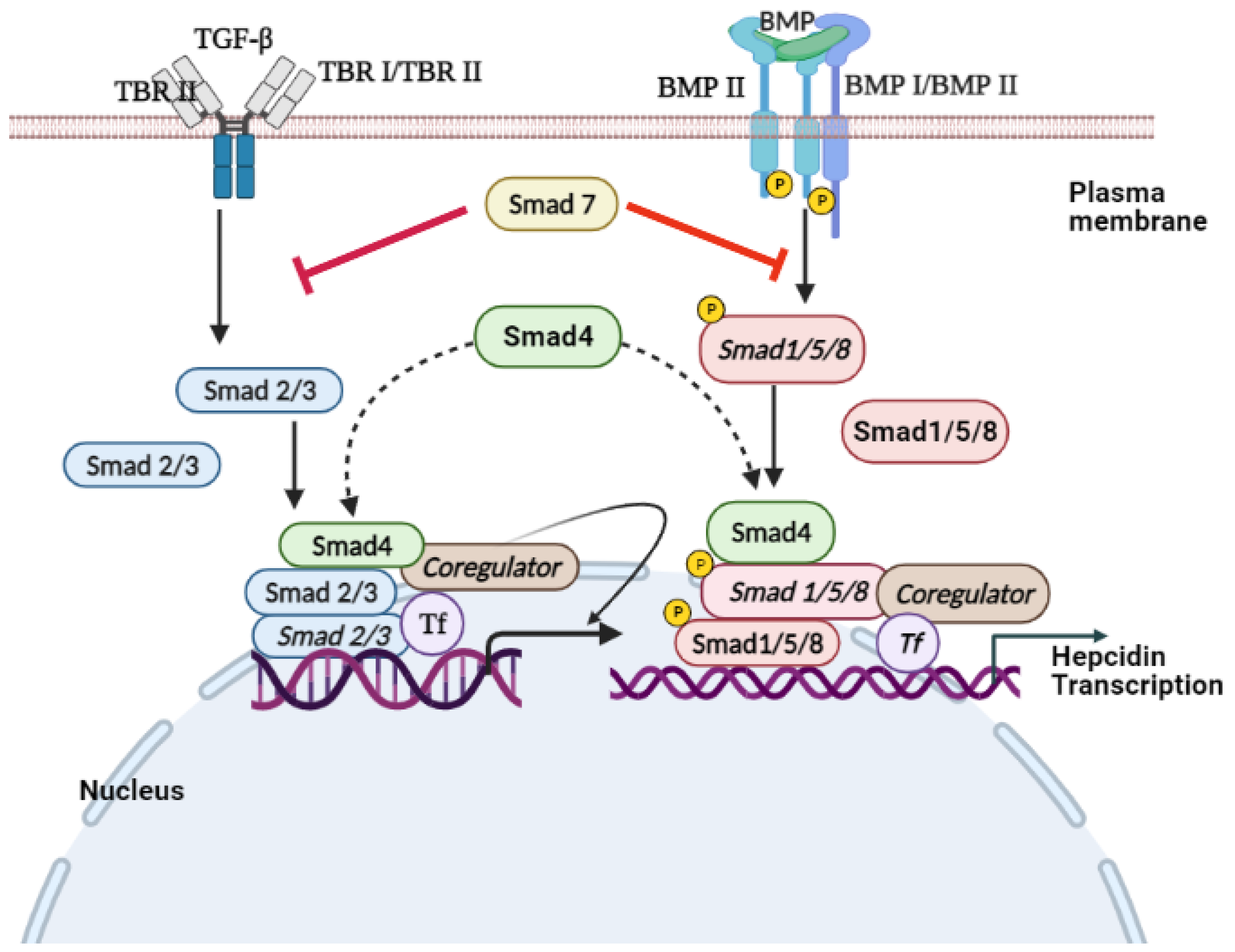
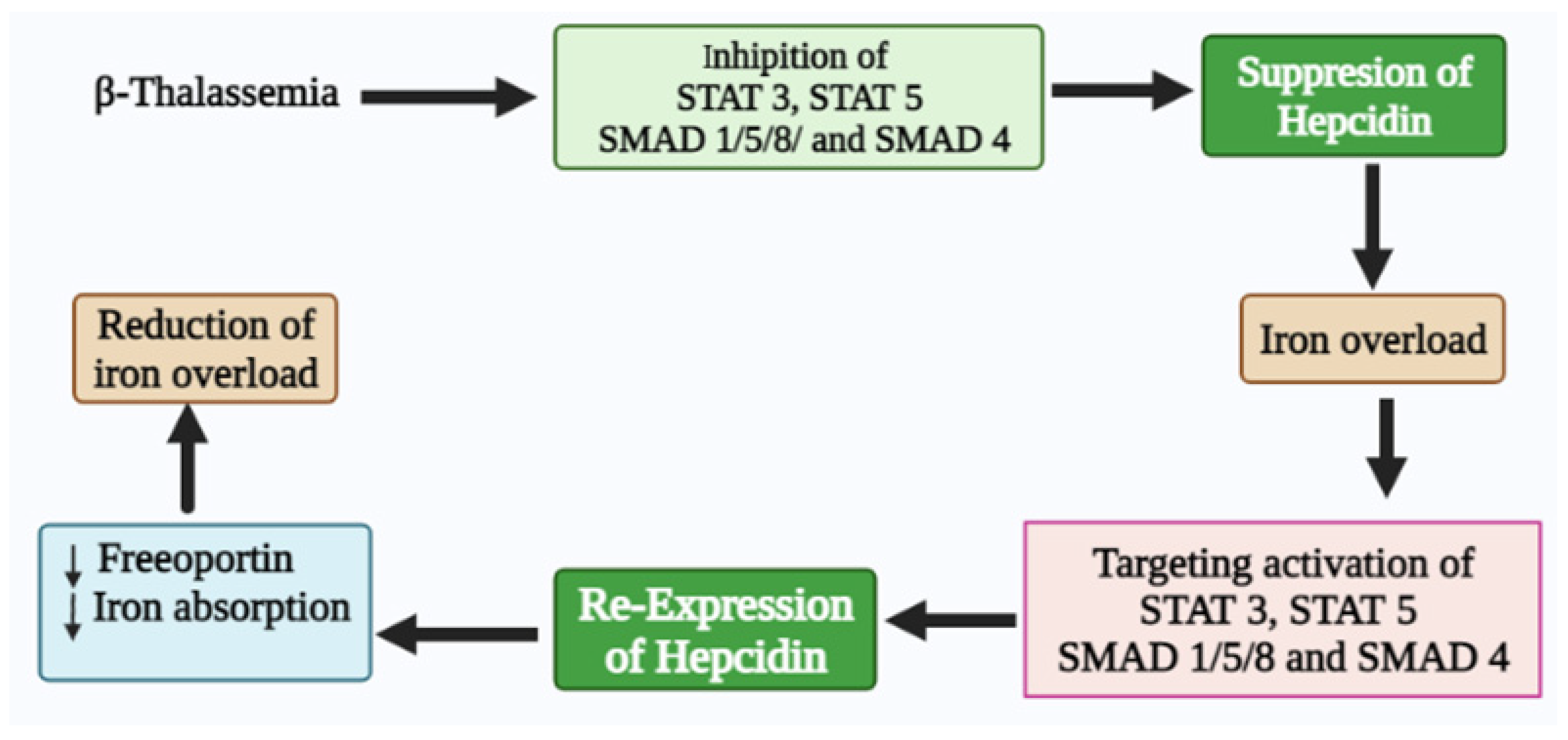
4. Conclusions
References
- Ayyash, H.; Sirdah, M. Hematological and biochemical evaluation of β-thalassemia major (βTM) patients in Gaza Strip: A cross-sectional study. IJHS 2018, 12, 18.
- Fibach, E.; Rachmilewitz, E.A. Pathophysiology and treatment of patients with beta-thalassemia—An update. F1000Research 2017, 6, 1–12.
- Aziz, N.; Taib, W.; Kharolazaman, N.; Ismail, I.; Al-Jamal, H.; Jamil, W.; Esa, E.; Ibrahim, H. Evidence of New Intragenic HBB Haplotypes Model for the Prediction of Beta-Thalassemia in the Malaysian Population. Res. Sq. 2021; preprint.
- Bender, M.; Yusuf, C.; Davis, T.; Dorley, M.C.; del Pilar Aguinaga, M.; Ingram, A.; Chan, M.S.; Ubaike, J.C.; Hassell, K.; Ojodu, J. Newborn Screening Practices and Alpha-Thalassemia Detection—United States, 2016. Morb. Mortal. Wkly. Rep. 2020, 69, 1269.
- Wong, L.P.; George, E.; Tan, J.-A.M.A. Public perceptions and attitudes toward thalassaemia: Influencing factors in a multi-racial population. BMC Public Health 2011, 11, 193.
- Galanello, R.; Origa, R. Beta-thalassemia. Orphanet J. Rare Dis. 2010, 5, 11.
- Thein, S.L. Molecular basis of β thalassemia and potential therapeutic targets. Blood Cell Mol. Dis. 2018, 70, 54–65.
- Abdullah, U.Y.; Ibrahim, H.M.; Mahmud, N.B.; Salleh, M.Z.; Teh, L.K.; Noorizhab, M.N.F.b.; Zilfalil, B.A.; Jassim, H.M.; Wilairat, P.; Fucharoen, S. Genotype-Phenotype Correlation of β-Thalassemia in Malaysian Population: Toward Effective Genetic Counseling. Hemoglobin 2020, 44, 184–189.
- Pani, K.; Sharma, S.; Murari, M.; Yadav, M.; Phadke, S.; Agarwal, S. Clinico-hematological Profile of Hb E-β Thalassemia-Prospective Analysis in a tertiary Care Centre. J. Assoc. Phys. India 2018, 66, 42–45.
- Fucharoen, S.; Weatherall, D.J. The hemoglobin E thalassemias. Cold Spring Harb. Perspect. Med 2012, 2, a011734.
- Olivieri, N.F.; Pakbaz, Z.; Vichinsky, E. Hb E/beta-thalassaemia: A common & clinically diverse disorder. Indian J. Med. Res. 2011, 134, 522.
- Adamsky, K.; Weizer, O.; Amariglio, N.; Breda, L.; Harmelin, A.; Rivella, S.; Rachmilewitz, E.; Rechavi, G. Decreased hepcidin mRNA expression in thalassemic mice. Br. J. Hematol. 2004, 124, 123–124.
- Saad, H.K.M.; Taib, W.R.W.; Ismail, I.; Johan, M.F.; Al-Wajeeh, A.S.; Al-jamal, H.A.N. Reduced hepcidin expression enhances iron overload in patients with HbE/β-thalassemia: A comparative cross-sectional study. Exp. Ther. Med. 2021, 22, 1–8.
- Tanno, T.; Bhanu, N.V.; Oneal, P.A.; Goh, S.-H.; Staker, P.; Lee, Y.T.; Moroney, J.W.; Reed, C.H.; Luban, N.L.; Wang, R.-H. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat. Med. 2007, 13, 1096–1101.
- Kattamis, A.; Papassotiriou, I.; Palaiologou, D.; Apostolakou, F.; Galani, A.; Ladis, V.; Sakellaropoulos, N.; Papanikolaou, G. The effects of erythropoetic activity and iron burden on hepcidin expression in patients with thalassemia major. Haematologica 2006, 91, 809–812.
- Jones, E.; Pasricha, S.-R.; Allen, A.; Evans, P.; Fisher, C.A.; Wray, K.; Premawardhena, A.; Bandara, D.; Perera, A.; Webster, C. Hepcidin is suppressed by erythropoiesis in hemoglobin E β-thalassemia and β-thalassemia trait. Blood J. Am. Soc. Hematol. 2015, 125, 873–880.
- Pak, M.; Lopez, M.A.; Gabayan, V.; Ganz, T.; Rivera, S. Suppression of hepcidin during anemia requires erythropoietic activity. Blood 2006, 108, 3730–3735.
- Weizer-Stern, O.; Adamsky, K.; Amariglio, N.; Levin, C.; Koren, A.; Breuer, W.; Rachmilewitz, E.; Breda, L.; Rivella, S.; Ioav Cabantchik, Z. Downregulation of hepcidin and haemojuvelin expression in the hepatocyte cell-line HepG2 induced by thalassaemic sera. Br. J. Hematol. 2006, 135, 129–138.
- Tanno, T.; Porayette, P.; Sripichai, O.; Noh, S.-J.; Byrnes, C.; Bhupatiraju, A.; Lee, Y.T.; Goodnough, J.B.; Harandi, O.; Ganz, T. Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood J. Am. Soc. Hematol. 2009, 114, 181–186.
- Kautz, L.; Jung, G.; Du, X.; Gabayan, V.; Chapman, J.; Nasoff, M.; Nemeth, E.; Ganz, T. Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of β-thalassemia. Blood J. Am. Soc. Hematol. 2015, 126, 2031–2037.
- Gardenghi, S.; Marongiu, M.F.; Ramos, P.; Guy, E.; Breda, L.; Chadburn, A.; Liu, Y.; Amariglio, N.; Rechavi, G.; Rachmilewitz, E.A. Ineffective erythropoiesis in β-thalassemia is characterized by increased iron absorption mediated by down-regulation of hepcidin and up-regulation of ferroportin. Blood J. Am. Soc. Hematol. 2007, 109, 5027–5035.
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A red carpet for iron metabolism. Cell J. 2017, 168, 344–361.
- Pinto, J.P.; Ribeiro, S.; Pontes, H.; Thowfeequ, S.; Tosh, D.; Carvalho, F.; Porto, G. Erythropoietin mediates hepcidin expression in hepatocytes through EPOR signaling and regulation of C/EBPα. Blood J. Am. Soc. Hematol. 2008, 111, 5727–5733.
- Liu, Q.; Davidoff, O.; Niss, K.; Haase, V.H. Hypoxia-inducible factor regulates hepcidin via erythropoietin-induced erythropoiesis. J. Clin. Investig. 2012, 122, 4635–4644.
- Jiang, F.; Yu, W.-J.; Wang, X.-H.; Tang, Y.-T.; Guo, L.; Jiao, X.-Y. Regulation of hepcidin through GDF-15 in cancer-related anemia. Clin. Chim. Acta 2014, 428, 14–19.
- YAŞAR, C.A.; Tarkun, P.; Ateşoğlu, E.B.; Eraldemir, F.C.; Özsoy, Ö.D.; Demirsoy, E.T.; Mehtap, Ö.; Gedük, A.; Hacihanifioğlu, A. The role of hepcidin, GDF15, and mitoferrin-1 in iron metabolism of polycythemia vera and essential thrombocytosis patients. Turk. J. Med. Sci. 2019, 49, 74–80.
- Osada, M.; Park, H.L.; Park, M.J.; Liu, J.-W.; Wu, G.; Trink, B.; Sidransky, D. A p53-type response element in the GDF15 promoter confers high specificity for p53 activation. Biochem. Biophys. Res. Commun. 2007, 354, 913–918.
- Zimmers, T.A.; Jin, X.; Hsiao, E.C.; McGrath, S.A.; Esquela, A.F.; Koniaris, L.G. Growth differentiation factor-15/macrophage inhibitory cytokine-1 induction after kidney and lung injury. Shock 2005, 23, 543–548.
- Jenkitkasemwong, S.; Broderius, M.; Nam, H.; Prohaska, J.R.; Knutson, M.D. Anemic copper-deficient rats, but not mice, display low hepcidin expression and high ferroportin levels. J. Nutr. 2010, 140, 723–730.
- Finkenstedt, A.; Widschwendter, A.; Brasse-Lagnel, C.; Theurl, I.; Hubalek, M.; Dieplinger, H.; Tselepis, C.; Ward, D.; Vogel, W.; Zoller, H. Hepcidin is correlated to soluble hemojuvelin but not to increased GDF15 during pregnancy. Blood Cell Mol. Dis. 2012, 48, 233–237.
- Vokurka, M.; Krijt, J.; Šulc, K.; Nečas, E. Hepcidin mRNA levels in mouse liver respond to inhibition of erythropoiesis. Physiol. Res. 2006, 55, 667–674.
- Kautz, L.; Jung, G.; Valore, E.V.; Rivella, S.; Nemeth, E.; Ganz, T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet. 2014, 46, 678–684.
- Patel, N.; Varghese, J.; Masaratana, P.; Latunde-Dada, G.O.; Jacob, M.; Simpson, R.J.; McKie, A.T. The transcription factor ATOH 8 is regulated by erythropoietic activity and regulates HAMP transcription and cellular pSMAD 1, 5, 8 levels. Br. J. Hematol. 2014, 164, 586–596.
- Wrighting, D.M.; Andrews, N.C. Interleukin-6 induces hepcidin expression through STAT3. Blood 2006, 108, 3204–3209.
- Bayele, H.K.; Srai, S.K.S. Genetic variation in hepcidin expression and its implications for phenotypic differences in iron metabolism. Haematologica 2009, 94, 1185.
- Andriopoulos Jr, B.; Corradini, E.; Xia, Y.; Faasse, S.A.; Chen, S.; Grgurevic, L.; Knutson, M.D.; Pietrangelo, A.; Vukicevic, S.; Lin, H.Y. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat. Genet. 2009, 41, 482–487.
- Bridle, K.R.; Frazer, D.M.; Wilkins, S.J.; Dixon, J.L.; Purdie, D.M.; Crawford, D.H.; Subramaniam, V.N.; Powell, L.W.; Anderson, G.J.; Ramm, G.A. Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homoeostasis. Lancet 2003, 361, 669–673.
- Zhou, X.Y.; Tomatsu, S.; Fleming, R.E.; Parkkila, S.; Waheed, A.; Jiang, J.; Fei, Y.; Brunt, E.M.; Ruddy, D.A.; Prass, C.E. HFE gene knockout produces mouse model of hereditary hemochromatosis. Proc. Natl. Acad. Sci. USA 1998, 95, 2492–2497.
- Feder, J.; Gnirke, A.; Thomas, W.; Tsuchihashi, Z.; Ruddy, D.; Basava, A.; Dormishian, F.; Domingo, R.; Ellis, M.; Fullan, A. A novel MHC class I–like gene is mutated in patients with hereditary haemochromatosis. Nat. Genet. 1996, 13, 399–408.
- Schmidt, P.J.; Toran, P.T.; Giannetti, A.M.; Bjorkman, P.J.; Andrews, N.C. The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression. Cell Metab. 2008, 7, 205–214.
- Lebrón, J.A.; Bennett, M.J.; Vaughn, D.E.; Chirino, A.J.; Snow, P.M.; Mintier, G.A.; Feder, J.N.; Bjorkman, P.J. Crystal structure of the hemochromatosis protein HFE and characterization of its interaction with transferrin receptor. Cell J. 1998, 93, 111–123.
- Ramey, G.; Deschemin, J.-C.; Vaulont, S. Cross-talk between the mitogen activated protein kinase and bone morphogenetic protein/hemojuvelin pathways is required for the induction of hepcidin by holotransferrin in primary mouse hepatocytes. Haematologica 2009, 94, 765–772.
- Poggi, M.; Sorrentino, F.; Pugliese, P.; Smacchia, M.P.; Daniele, C.; Equitani, F.; Terlizzi, F.; Guitarrini, M.R.; Monti, S.; Maffei, L. Longitudinal changes of endocrine and bone disease in adults with β-thalassemia major receiving different iron chelators over 5 years. Ann. Hematol. 2016, 95, 757–763.
- Farmaki, K.; Tzoumari, I.; Pappa, C. Oral chelators in transfusion-dependent thalassemia major patients may prevent or reverse iron overload complications. Blood Cell Mol. Dis. 2011, 47, 33–40.
- Poggiali, E.; Cassinerio, E.; Zanaboni, L.; Cappellini, M.D. An update on iron chelation therapy. Blood Transfus. 2012, 10, 411.
- Schmidt, P.J.; Racie, T.; Westerman, M.; Fitzgerald, K.; Butler, J.S.; Fleming, M.D. Combination therapy with a T mprss6 RNA i-therapeutic and the oral iron chelator deferiprone additively diminishes secondary iron overload in a mouse model of β-thalassemia intermedia. Am. J. Hematol. 2015, 90, 310–313.
- Sripichai, O.; Munkongdee, T.; Kumkhaek, C.; Svasti, S.; Winichagoon, P.; Fucharoen, S. Coinheritance of the different copy numbers of α-globin gene modifies severity of β-thalassemia/Hb E disease. Ann. Hematol. 2008, 87, 375–379.
- Lucarelli, G.; Isgrò, A.; Sodani, P.; Gaziev, J. Hematopoietic stem cell transplantation in thalassemia and sickle cell anemia. Cold Spring Harb. Perspect. Med 2012, 2, a011825.
- Camaschella, C.; Nai, A. Ineffective erythropoiesis and regulation of iron status in iron loading anaemias. Br. J. Hematol. 2016, 172, 512–523.
- Gardenghi, S.; Ramos, P.; Roy, C.N.; Andrews, N.C.; Nemeth, E.; An, X.; Narla, M.; Ginzburg, Y.; Rachmilewitz, E.A.; Giardina, P. Hepcidin as a Therapeutic Tool to Limit Iron Overload and Improve Anemia In β-Thalassemia. Blood 2010, 116, 1009.
- Wu, X.; Yung, L.-M.; Cheng, W.-H.; Yu, P.B.; Babitt, J.L.; Lin, H.Y.; Xia, Y. Hepcidin regulation by BMP signaling in macrophages is lipopolysaccharide dependent. PLoS ONE 2012, 7, e44622.
- Nai, A.; Lidonnici, M.R.; Rausa, M.; Mandelli, G.; Pagani, A.; Silvestri, L.; Ferrari, G.; Camaschella, C. The second transferrin receptor regulates red blood cell production in mice. Blood J. Am. Soc. Hematol. 2015, 125, 1170–1179.
- Finberg, K.E.; Heeney, M.M.; Campagna, D.R.; Aydınok, Y.; Pearson, H.A.; Hartman, K.R.; Mayo, M.M.; Samuel, S.M.; Strouse, J.J.; Markianos, K. Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA). Nat. Genet. 2008, 40, 569–571.
- Bou-Fakhredin, R.; Bazarbachi, A.-H.; Chaya, B.; Sleiman, J.; Cappellini, M.D.; Taher, A.T. Iron overload and chelation therapy in non-transfusion dependent thalassemia. Int. J. Mol. Sci. 2017, 18, 2778.
- Casu, C.; Aghajan, M.; Oikonomidou, P.R.; Guo, S.; Monia, B.P.; Rivella, S. Combination of Tmprss6-ASO and the iron chelator deferiprone improves erythropoiesis and reduces iron overload in a mouse model of beta-thalassemia intermedia. Haematologica 2016, 101, e8.
- Shi, Y.; Massagué, J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell J. 2003, 113, 685–700.
- Massagué, J. TGF-β signaling in development and disease. FEBS Lett. 2012, 586, 1833.
- Goh, J.B.; Wallace, D.F.; Hong, W.; Subramaniam, V.N. Endofin, a novel BMP-SMAD regulator of the iron-regulatory hormone, hepcidin. Sci. Rep. 2015, 5, 13986.
- Wang, R.-H.; Li, C.; Xu, X.; Zheng, Y.; Xiao, C.; Zerfas, P.; Cooperman, S.; Eckhaus, M.; Rouault, T.; Mishra, L. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005, 2, 399–409.
- Chen, J.; Chloupkova, M. Abnormal iron uptake and liver cancer. Cancer Biol. Ther. 2009, 8, 1699–1708.
- Mleczko-Sanecka, K.; Casanovas, G.; Ragab, A.; Breitkopf, K.; Müller, A.; Boutros, M.; Dooley, S.; Hentze, M.W.; Muckenthaler, M.U. SMAD7 controls iron metabolism as a potent inhibitor of hepcidin expression. Blood J. Am. Soc. Hematol. 2010, 115, 2657–2665.
- Spasić, M.V.; Sparla, R.; Mleczko-Sanecka, K.; Migas, M.C.; Breitkopf-Heinlein, K.; Dooley, S.; Vaulont, S.; Fleming, R.E.; Muckenthaler, M.U. Smad6 and Smad7 are co-regulated with hepcidin in mouse models of iron overload. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 76–84.
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGFβ activation. J. Cell Sci. 2003, 116, 217–224.
- Blobe, G.C.; Schiemann, W.P.; Lodish, H.F. Role of transforming growth factor β in human disease. N. Engl. J. Med. 2000, 342, 1350–1358.
- Massague, J. TGF-β signal transduction. Annu. Rev. Biochem. 1998, 67, 753–791.
- Salma, U.; Xue, M.; Ali Sheikh, M.S.; Guan, X.; Xu, B.; Zhang, A.; Huang, L.; Xu, D. Role of transforming growth factor-β1 and smads signaling pathway in intrauterine adhesion. Mediat. Inflamm. 2016, 2016, 4158287.
- Chen, Y.-G. Endocytic regulation of TGF-β signaling. Cell Res 2009, 19, 58–70.
- Di Guglielmo, G.M.; Le Roy, C.; Goodfellow, A.F.; Wrana, J.L. Distinct endocytic pathways regulate TGF-β receptor signalling and turnover. Nat. Cell Biol. 2003, 5, 410–421.
- Clarke, D.C.; Betterton, M.; Liu, X. Systems theory of Smad signalling. IEEE Proc. Syst. Biol. 2006, 153, 412–424.
- Schmierer, B.; Hill, C.S. Kinetic analysis of Smad nucleocytoplasmic shuttling reveals a mechanism for transforming growth factor β-dependent nuclear accumulation of Smads. Mol.Cell. Biol. 2005, 25, 9845–9858.
- Massagué, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810.

