Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
An, S. Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS). Encyclopedia. Available online: https://encyclopedia.pub/entry/18741 (accessed on 24 July 2026).
An S. Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS). Encyclopedia. Available at: https://encyclopedia.pub/entry/18741. Accessed July 24, 2026.
An, Seong. "Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS)" Encyclopedia, https://encyclopedia.pub/entry/18741 (accessed July 24, 2026).
An, S. (2022, January 25). Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS). In Encyclopedia. https://encyclopedia.pub/entry/18741
An, Seong. "Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS)." Encyclopedia. Web. 25 January, 2022.
Copy Citation
Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) is an early-onset neurodegenerative disease that was originally discovered in the population from the Charlevoix-Saguenay-Lac-Saint-Jean (CSLSJ) region in Quebec.
ARSACS
neurodegeneration
sacsin
1. Introduction
The sacsin gene (SACS) is located on chromosome 13 (13q12.12: chr13:23,288,689-23,433,763, GRCh38/hg38), with 145,075 bases, and is oriented in the minus strand of DNA (https://www.genecards.org/cgi-bin/carddisp.pl?gene=SACS accessed on 24 December 2021). SACS comprises 10 exons, with nine coding exons, and the 10th exon contains 11,487 base pairs, notable as the longest exon among vertebrates [1][2]. The SACS gene encodes a large 520-kDa multidomain protein of 4579 amino acids, called sacsin. It contains several different domains, including the ubiquitin-like (UBL) domain in the N-terminal region, three sacsin internal repeat (SIRPT or SRR) domains, the helical XPC-binding domain, a sacsin J-domain and the higher eukaryotes and prokaryotes nucleotide-binding (HEPN) domain in the C-terminal region [3]. Sacsin is expressed in several different tissues, with higher expression in the central nervous system or skin and lower expression in the pancreas and skeletal muscle. In the brain, sacsin expression is the highest in the motor system, including the cerebellum, granular system and in Purkinje cells [2].
SACS is associated with early-onset cerebellar ataxia due to mutation, called the spastic ataxia of Charlevoix-Saguenay (ARSACS), in an autosomal recessive pattern; it was first discovered in a population by the linkage disequilibrium studies conducted in the Charlevoix-Saguenay-Lac-Saint-Jean (CSLSJ) region in Quebec [4][5]. Based on several relatives with ataxia syndrome, the founder effect was present in the French Canadian population. Since this family immigrated to French Canada in the 17th century from the Perche region, it is possible that the disease was present in France, although similar cases may remain unrecognized. Other cases of SACS ataxia in non-Quebec populations, such as in Italy, Japan, Spain, Tunisia or Turkey, were discovered [6][7][8].
ARSACS is associated with ataxia, dysarthria, nystagmus, spasticity, distal muscle wasting and deformities in fingers or feet [6][7]. Affected patients show slow progression of spastic ataxia, which may affect all four limbs. Patients also experience a loss of muscle tissue (amyotrophy) and language impairment (their speech became slurred). Ocular movements may also be impaired [6][7]. Disease may occur during childhood (lower limb ataxia), but their intelligence may not be impaired. Cerebellar signs and pes cavus appear until patients reach their 20 s. Other characteristics may also be possible, such as retinal nerve fiber hypermyelination [8][9][10]. The first characteristics of the disease may be gait initiation, which can be noticed when children start to walk (around 12–18 months of age). Childhood ataxia and spasticity may be prominent as well. In early adulthood (20 s or 30 s), disease progression may be accelerated, and patients may lose the ability to walk by the age of 50 [9]. Bladder and bowel dysfunctions could appear in patients in their 50 s [10]. In addition, patients may show biochemical dysfunctions, such as impaired pyruvate oxidation, hyperbilirubinemia and low serum beta- or HDL lipoproteins [11]. In patients with ARSACS, non-phosphorylated neurofilaments (NFs) may occur in different neurons, including Purkinje cells or motor neurons in the cortex. The cultured motor neurons of SACS knockout mouse embryos presented abnormal neurofilament rearrangements [12][13]. Abnormal mitochondrial functions were also detected, with lower mitochondrial motility and elongation. Since NFs could impact cytoskeletal organization, the alteration of NFs in patients with ARSACS may present mitochondrial impairments, resulting in elevated cellular vulnerability [12][13]. In this review, mutations in the SACS gene in patients with ARSACS and the potential involvement of sacsin in other forms of neurodegeneration are discussed.
2. Potential Involvement of SACS in Other Neurodegenerative Diseases
Currently, SACS has only been related to ARSACS. According to several studies on non-functional sacsin or sacsin knockout in cell lines or mice, sacsin is involved in various cellular roles, such as chaperon functions, mitochondrial mechanisms, microtubule filament control and cell adhesion [14]. It may not be ruled out that sacsin could exert an impact, directly or indirectly, in other types of neurodegenerative diseases (especially ataxias) than ARSACS. Since sacsin could interact with Hsp70 and ubiquitin proteasomes, its association may be involved in the defensive mechanisms against abnormal protein aggregations. When sacsin expression was attenuated, higher toxicity of repeat expansion was observed in comparison to normal sacsin expression. With Hsp70, sacsin may regulate the processing of ataxin-1 with polyglutamate expansion, especially the aberrant ataxin-1 degradation for protection against spinocerebellar ataxia-1 [2]. A putative association between sacsin and ataxin-3 was reported, where the N-terminal UbL domain of sacsin could directly interact with proteasomes. Since ataxin-3 could also interact with proteasomes, sacsin could affect the pathogenic mechanisms of ataxin-3 dysfunctions [15].
Interestingly, newly discovered SACS mutations in suspected patients with CMT-like neuropathy and atypical disease phenotypes suggest a potential pathological overlap between ARSACS and CMT [4].
The involvement of the SACS gene in other neurodegenerative diseases, such as AD, PD, ALS or CJD, has not been reported yet. Sacsin is closely involved in controlling the mitochondrial functions and dynamics. Sacsin dysfunctions have been associated with impairment of mitochondrial morphology, dynamics, organization and dysfunctions in Drp1, and could cause synaptic dysfunctions and loss of Purkinje cells [16][17]. In addition, the interactions between Drp1 and sacsin for proper mitochondrial functions, and Drp dysfunctions in the imbalance of mitochondrial fusion or fission, could also indicate the involvement of SACS mutations in AD, ALS, MS or PD. Altered Drp1 expression could cause the overexpression of amyloid beta, huntingtin or alpha synuclein. By interacting with Drp1, sacsin may influence the expression of different neurodegenerative-disease-related proteins indirectly. Defects in mitochondrial dynamics in other neurodegenerative disorders could be a common pathway associated with ARSACS and with SACS mutations. Hence, the contribution of sacsin in different neurodegenerative diseases could be hypothesized [18][19][20].
An additional putative common pathway between sacsin and other neurodegenerative diseases could be through its chaperon function. Sacsin contains homologous sequences with Hsp90. In addition, sacsin could interact with Hsp70 to exert several neuroprotective mechanisms [21]. Hsp proteins may play as a key role in protein folding and protect neurons against various protein-folding-related diseases. Impairment of Hsps and other chaperons could result in elevated oxidative stress and mitochondrial dysfunctions. Overexpressed Hsp70 was reported in AD mouse models, suggesting enhanced protective mechanisms by reducing APP cleavage and amyloid peptide production. Hsp70 could also enhance the transport of Tau protein and amyloid oligomers into proteasomes [22]. Hsp70 seemed to play an essential role in protecting against prion misfolding and aggregation [23]. In Hsp70 knockout mice, prion propagation and toxicity was accelerated [24]. Lastly, Hsp70 could block alpha synuclein oligomerization through a noncanonical site in the C-terminal domain to exert a protective function against PD and other synucleopathies [24]. These studies suggest that sacsin may contribute either directly or indirectly to neuroprotection against non-ataxia-related diseases. Figure 1 summarizes the possible common pathways between sacsin and other neurodegenerative diseases.
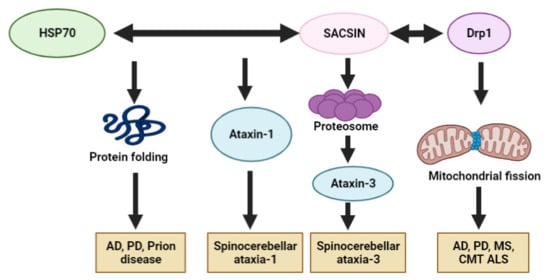
Figure 1. Possible disease mechanisms of sacsin protein in other neurodegenerative diseases: AD, PD, MS, CMT, ALS, SCA and CJD.
Additional evidence of an association involving common pathways between sacsin and other forms of neurodegenerative diseases was published by Morani et al. (2020) [25]. Their study analyzed proteomic data from ARSACS mouse models and isolated cells from ARSACS patients using SomaLogic technology. Several dysregulated pathways and differentially expressed proteins (DEPs) were found to be associated with neuroinflammation, synaptogenesis or cell engulfment. Several DEPs were found, which were involved in other neurodegenerative diseases: AD, PD, dementia with Lewy bodies and spastic paraplegia. Significant DEPs in ARSACS models were ephrins (EFNB2, EPHA3 and EPHB2), SNCA, APOE, ICAM5, SPHK1 and/or STUB1. This result points to the possibility of shared pathways between ARSACS and other neurodegenerative diseases. Hence, DEPs may act as risk factors or risk modifiers for ARSACS disease progression. Nevertheless, sacsin dysfunctions may alter the expression of ARSACS-related genes/proteins and may impact the pathological mechanisms of other neurodegenerative diseases, including AD, PD, ALS and CJD [25].
3. ARSACS Diagnosis and Potential Therapeutics
Genetic testing is required for the specific diagnosis of ARSACS. The standard Sanger sequencing would be challenging due to the size of the SACS gene. Hence, next-generation sequencing techniques would be the optimal approach in the discovery of novel causative genes or mutations in the SACS gene [26]. Additional biomarkers would be needed to enhance the disease’s diagnosis in combination with imaging biomarkers, such as brain and retinal imaging, in ARSACS diagnosis. Interestingly, “bithalamic stripes” detected by MRI imaging or retinal nerve fiber layer thickening could be an additional useful diagnostic biomarker of the disease [27].
Retinal nerve thickening was reported in several ARSACS patients. Recently, optical coherence tomography (OCT) was suggested to represent a significant diagnostic tool for investigating visual impairments and diseases in retina or neuropathy, as a non-invasive and cost-effective test. Hence, OCT could observe retinal nerve fiber thickening in patients with ARSACS [28][29]. Parkinson et al. (2018) performed OCT on patients with different types of ataxia (191) and controls (101). Retinal nerve fiber thickening was present only in ARSACS cases, and not in controls or other kinds of ataxia, such as Friedrich ataxia or spinocerebellar ataxia. This study proposed a cut-off value of 119 μm for the average retinal nerve fiber thickness. This value provided high specificity and sensitivity (100% and 99.4%, respectively) among ataxia patients [28]. Since the retina may be an excellent source of potential surrogate biomarkers in ARSACS, Rezende Filho et al. (2021) performed fundoscopy (another simple cost-effective technique) and OCT on patients with ARSACS and other forms of ataxia (spinocerebellar ataxia, autosomal recessive cerebellar ataxia, hereditary spastic paraplegia). The investigated retinal nerve fiber thickening in ARSACS by fundoscopy provided false negative data, suggesting that this method has lower sensitivity than OCT [30].
ARSACS patients presented other types of retinal abnormalities in comparison to control patients or those with other forms of ataxia. All ARSACS patients presented foveal hypoplasia, in addition to other impairments, such as retinal hyperplasia, sawtooth appearance or papillomacular fold. The above results suggest that monitoring neurophysiological abnormalities could yield promising biomarkers for ARSACS diagnosis [28][30], such as nerve conduction or nerve ultrasonography. Nerve enlargement and peripheral demyelination may be useful biomarkers in ARSACS [31][25]. Since the typical imaging biomarkers could be missing in some cases of ARSACS, Pilliod et al. analyzed 321 diagnosed patients with spinocerebellar degeneration from the SPATAX (http://spatax.wordpress.com accessed on 23 September 2021) database. They also collected fibroblast samples from skin biopsies in 11 ARSACS patients and 8 controls in order to perform mitochondrial morphology analyses, which indicated that mitochondrial abnormalities, such as bulbed mitochondria, were common among ARSACS patients. In addition, mitochondrial mass, oxygen consumption and the ratio of mitochondrial DNA/nuclear DNA were reduced among ARSACS patients. These anomalies in the mitochondrial network may be a useful diagnostic and prognostic biomarker to predict the pathogenicity of ARSACS [31]. Since spasticity is one of the typical key features, with reduced movement and coordination in patients with ARSACS, Lessard et al. performed the Lower Extremity Motor Coordination Test (LEMOCOT) for its possible usage in attempting diagnosis [32]. Analyzing pendulum oscillation amplitudes and their ratios using a wireless electro-goniometer provided information on the degree of spasticity in ARSACS patients. This device could effectively measure the evolution of spasticity in patients and provide an easy tool to compare the data from the pendulum between patients and unaffected individuals. Hence, this method would be an easy, rapid and cost-effective test for ARSACS diagnosis [33].
Since no therapy is currently available for ARSACS, the best disease management strategy is to mitigate the disease symptoms and improve the quality of the patient’s life. Positive associations were confirmed between fitness activities and the symptoms of neuromuscular/neurological diseases. An eight-week workout program, used by Audet et al. (2018), suggested that physical training may be beneficial for the fitness and functional capacity of ARSACS patients. Strengthened muscular and functional capacity was observed in patients who participated in the program. In addition, the regular workout improved patients’ ability to perform their daily activities. Furthermore, the reduced frequency of falls was notable. Regular training in patients with ARSACS could help them to retain and even regain their independence of movement [34]. Speech training was also effective to treat language impairments. Vogel et al. (2019) recruited ARSACS patients into a 4-week program with rater-blinded assessment of intelligibility. Although this was a preliminary study, the speech treatment improved the intelligibility of patients and enhanced the spontaneity of their speech [35].
Although no effective drug is available for ARSACS yet, certain candidate drugs may improve the quality of life of ARSACS patients. The main criterion for drug development is their capacity to cross the blood–brain barrier [36]. Docosahexaenoic acid (DHA), a dietary supplement, was investigated for its possible neuroprotective functions—namely, its anti-apoptosis, anti-inflammatory and anti-autophagic properties. Since DHA contains phospholipids, similar lipids to brain phospholipids, it was proposed in patients with ataxias with cerebellar and pyramidal involvement. Ricca et al. (2020) carried out a small clinical study in two ARSACS patients with SACS mutations. DHA was administrated orally for 20 months. Afterwards, investigators noticed stalled or slowed disease progression or the deterioration of clinical symptoms, with slight improvements in the functioning of the lower limbs and speech being reported, suggesting DHA as a safe and promising add-on therapy in patients with ARSACS and SACS mutations [37].
In addition, Idebenone (IDE), an analogue of coenzyme Q10, was successfully used to treat brain disorders with mitochondrial etiology by protecting against free radical toxicity. Since the solubility of IDE is low, many researchers have investigated it for the best drug delivery systems. Nanostructured lipid carriers (NLCs) loaded with IDE, with stability in water or cell media, and with successful penetration of the blood–brain barrier, were invented, representing a promising future approach in ARSACS therapy [36].
In sacsin knockout cells, the modulation of PTEN-FAK signaling could improve the ARSACS-related cellular dysfunctions, including microtubule organization, cell migration or adhesion [14]. PTEN has been verified as a negative regulator of focal adhesion. Hence, PTEN signal modulation has been suggested as a possible therapeutic target in ARSACS.
Since higher inductions of Hsp90 are observed in neurodegenerative diseases, a Hsp90 inhibitor was investigated for a possible reduction in disease progression and toxicity. A potential therapeutic candidate was KU-32, a Hsp90 inhibitor [38]. Hence, inhibiting Hsp90 was contemplated for its possible benefits in patients with ARSACS and different neurodegenerative disorders [39]. Ku-32 treatment improved the mitochondrial functions (electron transport, mitochondrial membrane potential) in cells among ARSACS patients [38].
References
- Takiyama, Y. Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Neuropathology 2006, 26, 368–375.
- Parfitt, D.A.; Michael, G.J.; Vermeulen, E.G.; Prodromou, N.V.; Webb, T.R.; Gallo, J.-M.; Cheetham, M.E.; Nicoll, W.S.; Blatch, G.L.; Chapple, J.P. The ataxia protein sacsin is a functional co-chaperone that protects against polyglutamine-expanded ataxin-1. Hum. Mol. Genet. 2009, 18, 1556–1565.
- Gentil, B.J.; Lai, G.-T.; Menade, M.; Larivière, R.; Minotti, S.; Gehring, K.; Chapple, J.-P.; Brais, B.; Durham, H.D. Sacsin, mutated in the ataxia ARSACS, regulates intermediate filament assembly and dynamics. FASEB J. 2019, 33, 2982–2994.
- Synofzik, M.; Soehn, A.S.; Gburek-Augustat, J.; Schicks, J.; Karle, K.N.; Schüle, R.; Haack, T.B.; Schöning, M.; Biskup, S.; Rudnik-Schöneborn, S.; et al. Autosomal recessive spastic ataxia of Charlevoix Saguenay (ARSACS): Expanding the genetic, clinical and imaging spectrum. Orphanet J. Rare Dis. 2013, 8, 41.
- Engert, J.; Bérubé, P.; Mercier, J.; Doré, C.; Lepage, P.; Ge, B.; Bouchard, J.-P.; Mathieu, J.; Melançon, S.B.; Schalling, M.; et al. ARSACS, a spastic ataxia common in northeastern Québec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat. Genet. 2000, 24, 120–125.
- De Braekeleer, M.; Giasson, F.; Mathieu, J.; Roy, M.; Bouchard, J.P.; Morgan, K. Genetic epidemiology of autosomal recessive spastic ataxia of Charlevoix-Saguenay in northeastern Quebec. Genet. Epidemiol. 1993, 10, 17–25.
- Heyer, E. Genetic consequences of differential demographic behaviour in the Saguenay region, Québec. Am. J. Phys. Anthr. 1995, 98, 1–11.
- Takiyama, Y. Sacsinopathies: Sacsin-related ataxia. Cerebellum 2007, 6, 353–359.
- Bouchard, J.-P.; Richter, A.; Mathieu, J.; Brunet, D.; Hudson, T.J.; Morgan, K.; Melançon, S.B. Autosomal recessive spastic ataxia of Charlevoix–Saguenay. Neuromuscul. Disord. 1998, 8, 474–479.
- Laberge, A.-M.; Michaud, J.; Richter, A.; Lemyre, E.; Lambert, M.; Brais, B.; Mitchell, G. Population history and its impact on medical genetics in Quebec. Clin. Genet. 2005, 68, 287–301.
- Bouhlal, Y.; Amouri, R.; El Euch-Fayeche, G.; Hentati, F. Autosomal recessive spastic ataxia of Charlevoix–Saguenay: An overview. Park. Relat. Disord. 2011, 17, 418–422.
- Larivière, R.; Gaudet, R.; Gentil, B.J.; Girard, M.; Conte, T.C.; Minotti, S.; Leclerc-Desaulniers, K.; Gehring, K.; McKinney, R.A.; Shoubridge, E.A.; et al. Sacs knockout mice present pathophysiological defects underlying autosomal recessive spastic ataxia of Charlevoix-Saguenay. Hum. Mol. Genet. 2014, 24, 727–739.
- Romano, A.; Tessa, A.; Barca, A.; Fattori, F.; Fulvia de Leva, M.; Terracciano, A.; Storelli, C.; Santorelli, F.M.; Verri, T. Comparative analysis and functional mapping of SACS mutations reveal novel insights into sacsin repeated architecture. Hum. Mutat. 2013, 34, 525–537.
- Romano, L.E.; Aw, W.Y.; Hixson, K.M.; Novoselova, T.V.; Havener, T.M.; Howell, S.; Hall, C.L.; Xing, L.; Beri, J.; Nethisinghe, S.; et al. The ataxia protein sacsin is required for integrin trafficking and synaptic organization. bioRxiv 2021.
- Burnett, B.; Li, F.; Pittman, R.N. The polyglutamine neurodegenerative protein ataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity. Hum. Mol. Genet. 2003, 12, 3195–3205.
- Girard, M.; Larivière, R.; Parfitt, D.A.; Deane, E.C.; Gaudet, R.; Nossova, N.; Blondeau, F.; Prenosil, G.; Vermeulen, E.G.M.; Duchen, M.; et al. Mitochondrial dysfunction and Purkinje cell loss in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Proc. Natl. Acad. Sci. USA 2012, 109, 1661–1666.
- Li, X.; Gehring, K. Structural studies of parkin and sacsin: Mitochondrial dynamics in neurodegenerative diseases. Mov. Disord. 2015, 30, 1610–1619.
- Feng, S.-T.; Wang, Z.-Z.; Yuan, Y.-H.; Wang, X.-L.; Sun, H.-M.; Chen, N.-H.; Zhang, Y. Dynamin-related protein 1: A protein critical for mitochondrial fission, mitophagy, and neuronal death in Parkinson’s disease. Pharmacol. Res. 2020, 151, 104553.
- Oliver, D.; Reddy, P.H. Dynamics of Dynamin-Related Protein 1 in Alzheimer’s Disease and Other Neurodegenerative Diseases. Cells 2019, 8, 961.
- Itoh, K.; Nakamura, K.; Iijima, M.; Sesaki, H. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 2013, 23, 64–71.
- Anderson, J.F.; Siller, E.; Barral, J.M. The Neurodegenerative-Disease-Related Protein Sacsin Is a Molecular Chaperone. J. Mol. Biol. 2011, 411, 870–880.
- Campanella, C.; Pace, A.; Caruso Bavisotto, C.; Marzullo, P.; Marino Gammazza, A.; Buscemi, S.; Palumbo Piccionello, A. Heat Shock Proteins in Alzheimer’s Disease: Role and Targeting. Int. J. Mol. Sci. 2018, 19, 2603.
- Mays, C.E.; Armijo, E.; Morales, R.; Kramm, C.; Flores, A.; Tiwari, A.; Bian, J.; Telling, G.C.; Pandita, T.K.; Hunt, C.R.; et al. Prion disease is accelerated in mice lacking stress-induced heat shock protein 70 (HSP70). J. Biol. Chem. 2019, 294, 13619–13628.
- Tao, J.; Berthet, A.; Citron, Y.R.; Tsiolaki, P.L.; Stanley, R.; Gestwicki, J.E.; Agard, D.A.; McConlogue, L. Hsp70 chaperone blocks α-synuclein oligomer formation via a novel engagement mechanism. J. Biol. Chem. 2021, 296, 100613.
- Morani, F.; Doccini, S.; Chiorino, G.; Fattori, F.; Galatolo, D.; Sciarrillo, E.; Gemignani, F.; Züchner, S.; Bertini, E.S.; Santorelli, F.M. Functional Network Profiles in ARSACS Disclosed by Aptamer-Based Proteomic Technology. Front. Neurol. 2020, 11, 603774.
- Zeng, H.; Tang, J.-G.; Yang, Y.-F.; Tan, Z.-P.; Tan, J.-Q. A Novel Homozygous SACS Mutation Identified by Whole-Exome Sequencing in a Consanguineous Family with Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay. Cytogenet. Genome Res. 2017, 152, 16–21.
- Kuchay, R.A.H.; Mir, Y.R.; Zeng, X.; Hassan, A.; Musarrat, J.; Parwez, I.; Kernstock, C.; Traschütz, A.; Synofzik, M. ARSACS as a Worldwide Disease: Novel SACS Mutations Identified in a Consanguineous Family from the Remote Tribal Jammu and Kashmir Region in India. Cerebellum 2019, 18, 807–812.
- Parkinson, M.H.; Bartmann, A.P.; Clayton, L.M.S.; Nethisinghe, S.; Pfundt, R.; Chapple, J.P.; Reilly, M.M.; Manji, H.; Wood, N.; Bremner, F.; et al. Optical coherence tomography in autosomal recessive spastic ataxia of Charlevoix-Saguenay. Brain 2018, 141, 989–999.
- Cho, H.; Lyoo, C.H.; Park, S.E.; Seo, Y.; Han, S.-H.; Han, J. Optical Coherence Tomography Findings Facilitate the Diagnosis of Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay. Korean J. Ophthalmol. 2021, 35, 330–331.
- Rezende Filho, F.M.; Bremner, F.; Pedroso, J.L.; de Andrade, J.B.C.; Marianelli, B.F.; Lourenço, C.M.; Marques-Júnior, W.; França, M.C.; Kok, F.; Sallum, J.M.; et al. Retinal architecture in autosomal recessive spastic ataxia of charlevoix-saguenay (ARSACS): Insights into disease pathogenesis and biomarkers. Mov. Disord. 2021, 36, 2027–2035.
- Pilliod, J.; Moutton, S.; Lavie, J.; Maurat, E.; Hubert, C.; Bellance, N.; Anheim, M.; Forlani, S.; Mochel, F.; N’Guyen, K.; et al. New practical definitions for the diagnosis of autosomal recessive spastic ataxia of Charlevoix-Saguenay. Ann. Neurol. 2015, 78, 871–886.
- Lessard, I.; Lavoie, C.; Côté, I.; Mathieu, J.; Brais, B.; Gagnon, C. Validity and reliability of the LEMOCOT in the adult ARSACS population: A measure of lower limb coordination. J. Neurol. Sci. 2017, 377, 193–1966.
- Bui, H.T.; Gagnon, C.; Audet, O.; Mathieu, J.; Leone, M. Measurement properties of a new wireless electrogoniometer for quantifying spasticity during the pendulum test in ARSACS patients. J. Neurol. Sci. 2017, 375, 181–185.
- Audet, O.; Bui, H.T.; Allisse, M.; Comtois, A.-S.; Leone, M. Assessment of the impact of an exercise program on the physical and functional capacity in patients with autosomal recessive spastic ataxia of Charlevoix-Saguenay: An exploratory study. Intractable Rare Dis. Res. 2018, 7, 164–171.
- Vogel, A.P.; Stoll, L.H.; Oettinger, A.; Rommel, N.; Kraus, E.-M.; Timmann, D.; Scott, D.; Atay, C.; Storey, E.; Schöls, L.; et al. Speech treatment improves dysarthria in multisystemic ataxia: A rater-blinded, controlled pilot-study in ARSACS. J. Neurol. 2019, 266, 1260–1266.
- Martinelli, C.; Battaglini, M.; Pucci, C.; Gioi, S.; Caracci, C.; Macaluso, G.; Doccini, S.; Santorelli, F.M.; Ciofani, G. Development of Nanostructured Lipid Carriers for the Delivery of Idebenone in Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay. ACS Omega 2020, 5, 12451–12466.
- Ricca, I.; Tessa, A.; Trovato, R.; Bacci, G.M.; Santorelli, F.M. Docosahexaenoic acid in ARSACS: Observations in two patients. BMC Neurol. 2020, 20, 215.
- Nethisinghe, S.; Abeti, R.; Kesavan, M.; Wigley, W.C.; Giunti, P. Hsp90 Inhibition: A Promising Therapeutic Approach for ARSACS. Int. J. Mol. Sci. 2021, 22, 11722.
- Zuehlke, A.D.; Moses, M.A.; Neckers, L. Heat shock protein 90: Its inhibition and function. Philos. Trans. R. Soc. B Biol. Sci. 2017, 373, 20160527.
More
Information
Subjects:
Health Care Sciences & Services
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Entry Collection:
Neurodegeneration
Revisions:
2 times
(View History)
Update Date:
25 Jan 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No