 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marco Maria D'Andrea | + 2767 word(s) | 2767 | 2020-08-19 08:11:16 | | | |
| 2 | Marco Maria D'Andrea | Meta information modification | 2767 | 2020-08-27 10:18:39 | | | | |
| 3 | Marco Maria D'Andrea | Meta information modification | 2767 | 2020-08-27 10:33:46 | | | | |
| 4 | Rita Xu | -572 word(s) | 2195 | 2020-08-27 10:37:08 | | | | |
| 5 | PASQUALE MARMO | Meta information modification | 2195 | 2020-08-27 19:10:37 | | | | |
| 6 | PASQUALE MARMO | Meta information modification | 2195 | 2020-08-28 10:46:43 | | |
Video Upload Options
Sphingomonas turrisvirgatae is a recently described species within the Sphingomonas genus. This species is endowed of agarolytic activity, a feature never reported before among sphingomonads. The capability of Sphingomonas sp. MCT13 to degrade agar, suggests that this strain could be potentially interesting for industrial applications in the field of complex carbohydrates degradation.
Sphingomonas is a very large and heterogeneous genus, and its huge biodiversity is responsible for the challenges of obtaining a reliable identification at the species level through conventional biochemical analyses. Indeed, bacteriophages specific for S. turrisvirgatae have been looked for, both as a rapid tool to identify and collect more isolates of this species, and for obtaining hints of its ecology.
vB_StuS_MMDA13 was isolated from a surface freshwater sample, obtained from a pond near Viterbo (Italy), by using the Sphingomonas turrisvirgatae MCT13T strain as host. The phage is lytic, belongs to the Siphoviridae family, and, as such, represents the first characterized lytic siphovirus able to infect a Sphingomonas species. vB_StuS_MMDA13 has a genome of ≈ 64 kb which encodes 89 potential proteins. A module for the biosynthesis of 7-deazaguanine derivatives, with a unique organization, is also present. The lysis module, which includes the endolysin, a holin/antiholin system and a Rz/Rz1 system, does not share significant similarities with other viral proteins in the databases. At the genome level vB_StuS_MMDA13 is loosely related to the Pseudomonas aeruginosa-infecting Nipunaviruses; however, both the 7-deazaguanine derivatives biosynthetic- and the lysis-modules are very different from these and other related bacteriophages. According to these features, we feel that vB_StuS_MMDA13 should be regarded as the type strain of a newly discovered genus within the Siphoviridae family, which we propose to name Ememdadecimater-like virus, after the short name of the first characterized phage of the genus.
1. Introduction
vB_StuS_MMDA13 is a lytic bacteriophage, belonging to the Siphoviridae family. It was isolated from a surface freshwater sample, in a pond near Viterbo (Italy), using the Sphingomonas turrisvirgatae MCT13T. [1] as host. The host range was determined on a total of 21 strains, including 8 Sphingomonas spp type strains (S. koreensis NBRC_16723, S. cynarae DSM 25525, S. hankookensis DSM 23329, S. insulae DSM 21792, S. pseudosanguinis DSM 19512, S. panni DSM 15761, S. soli DSM 18313 and S. naasensis DSM 100060), 6 type strains belonging to other genera of the Sphingomonadaceae family (Sphingobium yanoikuyae DSM 7462, Sphingobium chlorophenolicum DSM 7098, Novosphingobium aromaticivorans DSM 12444, Novosphingobium capsulatum DSM 30196, Sphingopyxis alaskensis DSM 13593 and Sphingopyxis macrogoltabida DSM 8826), Agrobacterium tumefaciens DSM 5172, Burkholderia cenocepacia J2315, and five Pseudomonas aeruginosa isolates. The results demonstrated that the lytic spectrum of this phage is restricted to S. turrisvirgatae MCT13T.
Electron microscopy revealed that vB_StuS_MMDA13 shows the morphological features of the B2 morphotype of the Siphoviridae family members [2], with an icosahedral elongated head (≈70x55 nm) and a long, flexible, helical, non-contractile and striated tail of about 140 nm ending with an apparent baseplate connected to short terminal fibers (Figure 1).
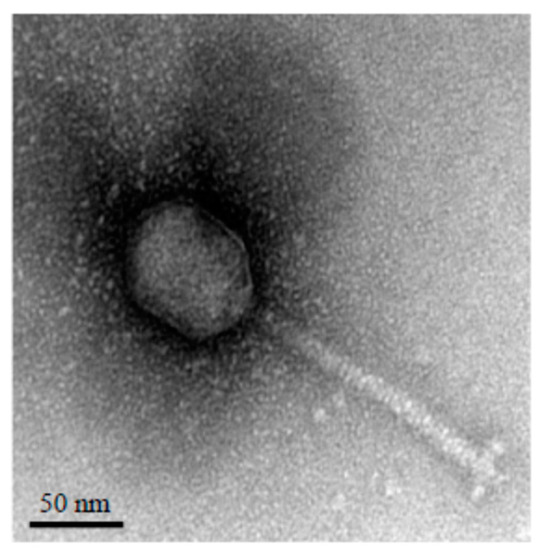
Figure 1. Transmission electron micrograph of the vB_StuS_MMDA13 phage negatively stained with uranyl acetate. The bar indicates 50 nm.
Phage particles are highly stable to both pH (range 3.0-11.0) and temperature variations (range 25-60 °C); the one-step growth curve showed long latency and rise periods, a feature observed also for other Siphoviridae infecting Alphaproteobacteria [3][4][5] which is mostly related to the slow growth of the host bacteria in liquid media. The computed average burst size was about 30 PFU per infected cell. Lisogeny was excluded both with mitomycin C treatment of three phage-insensitive colonies of S. turrisvirgatae MCT13T, obtained two days after a heavy infection, that revealed no production of phage particles, and by a PCR screening, using two different primers pairs targeting vB_StuS_MMDA13 specific genes, performed on the same phage-insensitive derivatives.
2. Genome Structure
The vB_StuS_MMDA13 genome is made up of a 63,743 bp long dsDNA encoding 89 predicted proteins and having a GC content of 59.3%, by far lower than that of S. turrisvirgatae MCT13T (65.3%). The nucleotide sequence of vB_StuS_MMDA13 has been deposited in the GenBank database under the accession number MN820898.
The coding percentage was equal to 94.3%, with a gene density of 1.40 genes/Kb. Most ORFs (76/89) started with ATG, 10 with GTG, 2 with TTG and one with ATC. A putative function could be assigned to 47 (52.8%) proteins by means of HHpred and HMMER analyses.
Sixty-one consecutive ORFs (38,382 bp; 60.2%) are encoded on the same DNA strand, and the remaining (21,208 bp; 33.3%) on the opposite strand. These two regions are separated by a 1,186 bp non coding segment having a GC content far lower than the rest of the phage genome (50.0 vs 59.3%), where four 12 bp imperfect direct repeats could be observed.
No tRNAs were found in the phage genome, suggesting that vB_StuS_MMDA13 cannot take over the host transcription/translation system, but relies on host tRNAs for the synthesis of phage proteins. This hypothesis is corroborated by the finding that for 16/18 (88.9%) aminoacids encoded by different triplets the same most abundant codon is shared between vB_StuS_MMDA13 and its host. As expected, no lysogeny associated elements were found.
3. Taxonomical Analysis
The taxonomic position of vB_StuS_MMDA13 has been tentatively evaluated by analyzing its terminase large subunit and major capsid protein (MCP) sequences. In both cases the obtained phylogenetic trees gave results highly consistent with the current ICTV taxonomy, but suggested also that vB_StuS_MMDA13 could not be assigned to any of known genera. vB_StuS_MMDA13 enters indeed in the same clade with Nipunavirus [6] but forms distinct nodes.
Similar results were obtained also by analyzing the full genome with both the Victor web-service, the OAT software (Figure 2) and the ViPTree web-service. According to the OAT analysis, vB_StuS_MMDA13 displayed a nucleotide identity ranging from 67% to 68% with the Nipunavirus classified by ICTV and with the unclassified phages JG012, JG054 and Quinobequin_P09 [7]. Interestingly, this analysis gave identical values when some members of different genera have been compared, e. g. Cajan and SE1, HdSG1 or JenK1.
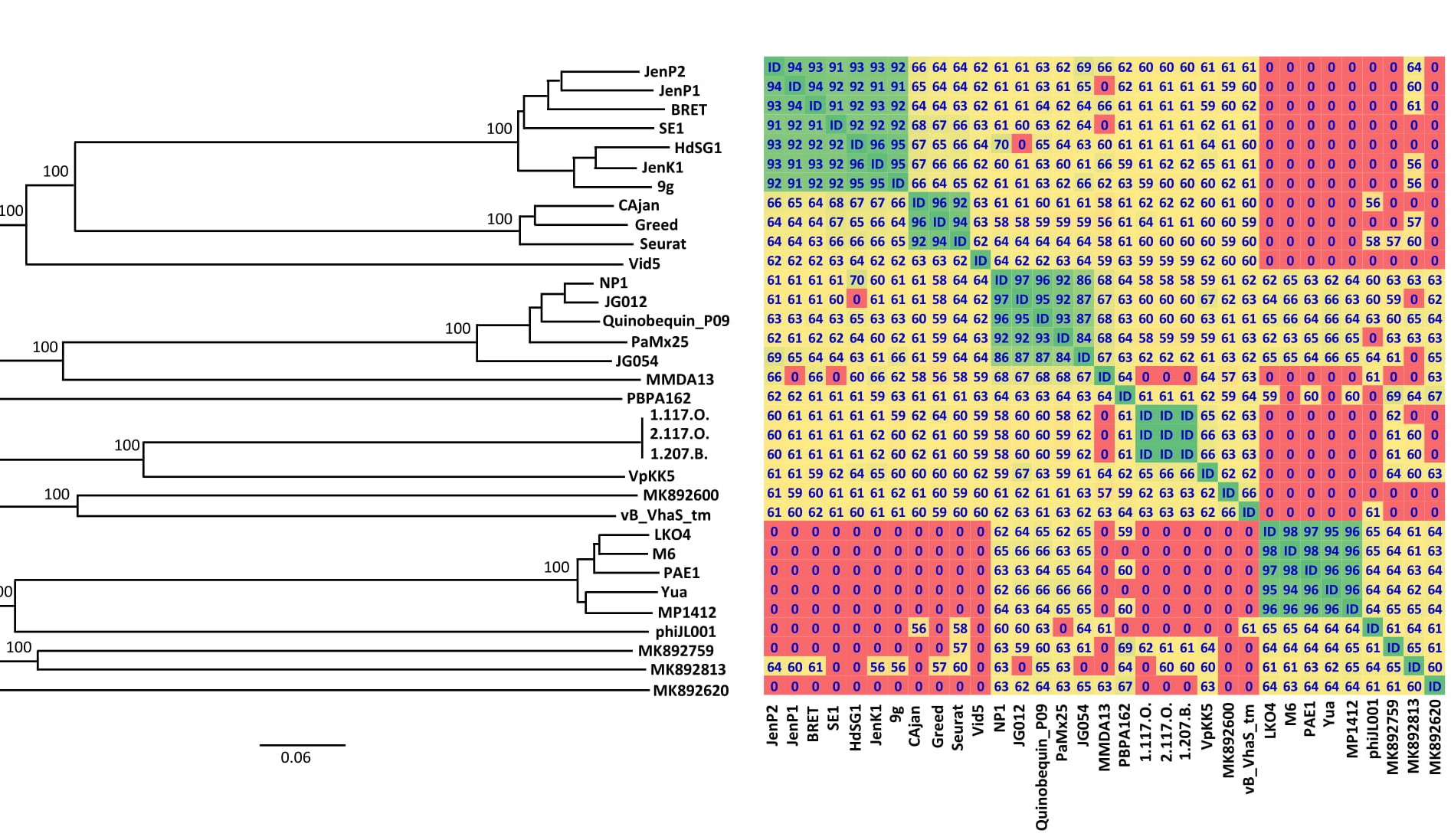
Figure 2. Phylogenomic Genome-BLAST Distance Phylogeny (GBDP) (on the left) and Orthologous Average Nucleotide Identity (OrthoANI) (on the right) analyses between vB_StuS_MMDA13 and the nearest members of Siphoviridae family. Only bootstrap values ≥ 95%, obtained by 100 replications in the GBDP analysis, are showed. Overall genomic sequence identity values are shown with different colours in a heatmap generated with the OrthoANI results. Cut-off is set at 50%: lower values are indicated as “0”. The following abbreviations are used: HdSG1, vB_EcoS_HdSG1; Vid5, vB_PagS_Vid5; MMDA13, vB_StuS_MMDA13; 1.117.O., Vibrio_phage_1.117.O._10N.261.45.E9; 2.117.O., Vibrio_phage_2.117.O._10N.261.45.E9; 1.207.B., Vibrio_phage_1.207.B._10N.222.51.C2; MK892600, Prokaryotic_dsDNA_virus_sp.; MK892759, Prokaryotic_dsDNA_virus_sp.; MK892813, Prokaryotic_dsDNA_virus_sp.; MK892620, Prokaryotic_dsDNA_virus_sp.
A CoreGenes analysis was performed to compare vB_StuS_MMDA13 with the type phages of the genera Nipuna-, Nonag-, Seurat- and Vidquinta-virus [6][7][8][9] and with all the unclassified bacteriophages selected according to the MCP similarities. The results showed that the highest percentage of shared ORFs (42.7 to 43.8%) was observed with the Nipunavirus/Quinobequin-P09 group.
4. Genome Organization
As commonly observed for most bacteriophages, the vB_StuS_MMDA13 genome is organized in modules, with genes related to different functions grouped in clusters. The transcription directions, on the genome, diverge from a 1,186 non-coding region interposed between gp53 and gp54 (Figure 3).
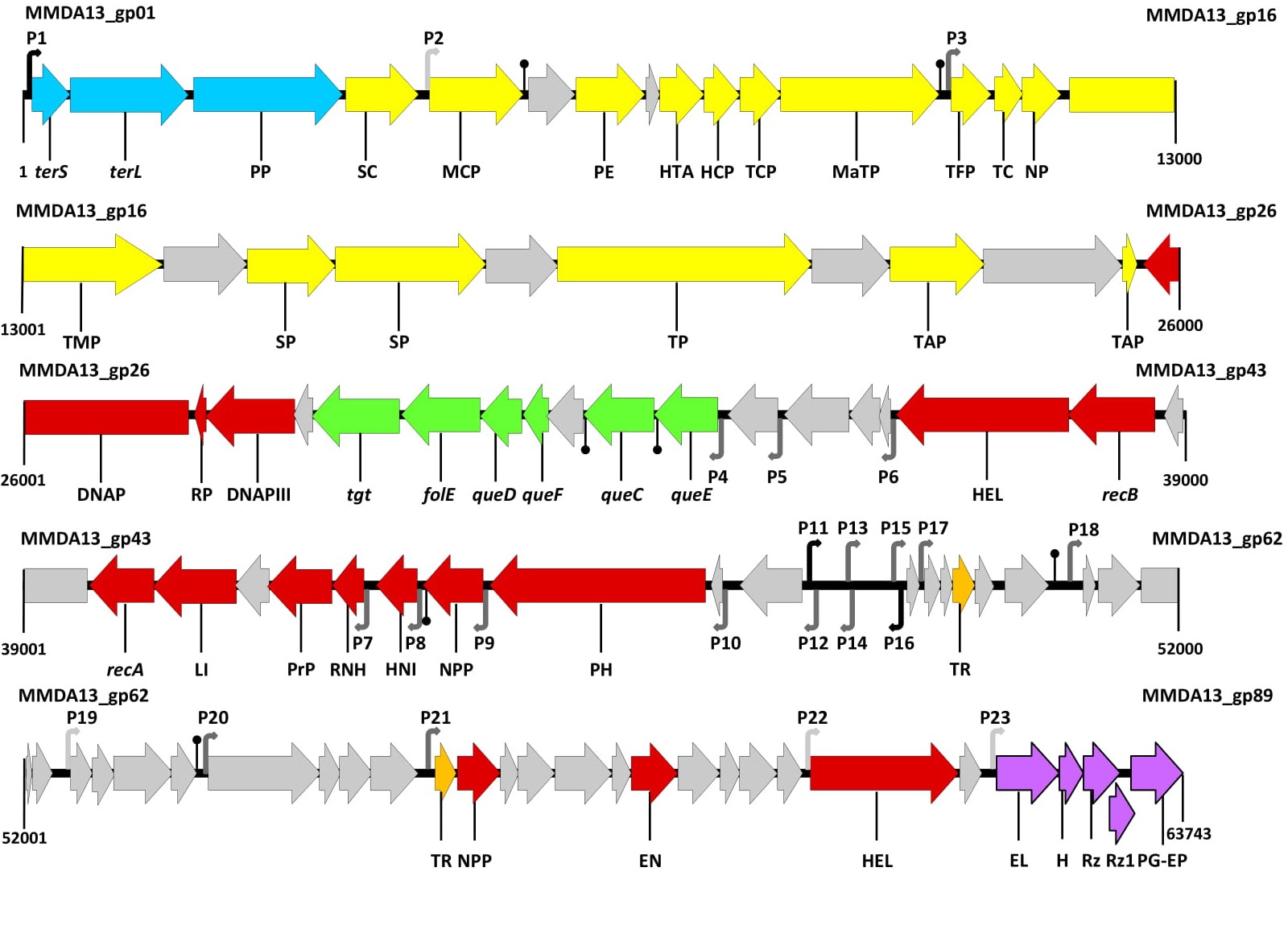
Figure 3. Functional genetic map of vB_StuS_MMDA13. Numbers indicate positions in the vB_StuS_MMDA13 genome (Accession number MN820898). Gene functions are assigned according to BLASTP and HHpred analyses. Colours of different functional modules are as follows: pale blue, packaging; yellow, morphogenesis; red, DNA replication/metabolism; green, modified nucleotides biosynthesis; orange, regulation; purple, lysis cassette. Open Reading Frames (ORFs) and a module of unknown function are reported in grey. ORFs names are as follows: terS, terminase small subunit; terL, terminase large subunit; PP, portal protein; SC, scaffold protein; MCP, major capsid protein; PE, peptidoglycan endopeptidase; HTA, head-tail adaptor; HCP, head-completion protein; TCP, tail completion protein; MaTP, main tail protein; TFP, tail fiber protein; TC, tail chaperonine; NP, neck protein; TMP, tail measure protein; SP, structural protein; TP, tail protein; TAP, tail assembly protein; DNAP, DNA polymerase; RP, DNA-directed RNA polymerase; DNAPIII, DNA polymerase III beta subunit; tgt, queuosine tRNA-ribosyltransferase; folE, GTP cyclohydrolase; queD, 6-carboxy-5,6,7,8-tetrahydropterin synthase; queF, 7-cyano-7-deazaguanine reductase; queC, 7-cyano-7-deazaguanine synthase; queE, 7-carboxy-7-deazaguanine synthase; HEL, helicase; recB, recB exonuclease; recA, recombinase A; LI, DNA ligase; PrP, primosomal protein; RNH, ribonuclease H; HNI, host-nuclease inhibitor; NPP, nucleotide pyrophosphohydrolase; PH, bifunctional primase helicase; TR, transcriptional regulator; EN, endonuclease; HEL, DNA helicase; EL, endolysin; H, holin/antiholin system; Rz, Rz protein; Rz1, Rz1 protein; PG-EP, peptidoglycan endopeptidase. Putative transcriptional promoters, detected with Bprom, are numbered (P1–P23) and shown as angled arrows according the transcription direction. The black, grey and pale grey arrows show putative promoters scoring more than 4, from 4 to 1, or less than 1, respectively. Putative rho-independent transcriptional terminators, detected with Genome2d, are depicted as vertical lines ending with a small black circle.
5. 7-deazaguanine derivatives module
A peculiar feature of vB_StuS_MMDA13 is the presence, within the DNA replication/nucleotide metabolism module, of a putative biosynthetic gene cluster for the synthesis of modified nucleotides. This cluster is made up of six ORFs, encoding the homologues of queuosine tRNA-ribosyltransferase (DpdA [10]), GTP cyclohydrolase, (FolE), 6-carboxy-5,6,7,8-tetrahydropterin synthase (QueD), 7-cyano-7-deazaguanine reductase (QueF-like), 7-cyano-7-deazaguanine synthase (QueC) and the organic radical activating enzyme, (QueE).
The organization of this cluster is unique due to the contemporary lack of a glutamine amidotransferase (GAT) domain within the QueC encoding ORF, and the presence of the QueF-like archaeosine synthase (Gp33), which provides a separate amidotransferase function. These data suggest that vB_StuS_MMDA13 belongs to the first group proposed by Hutinet et al. [7], and that it is likely able to modify its own DNA with archaeosine.
In Figure 4, the vB_StuS_MMDA13 nucleotide modification gene cluster is depicted and compared with those of type phages of related viral genera 9g, Vid5, Seurat, NP1 and Quinobequin-P09 [6][7][8][9].
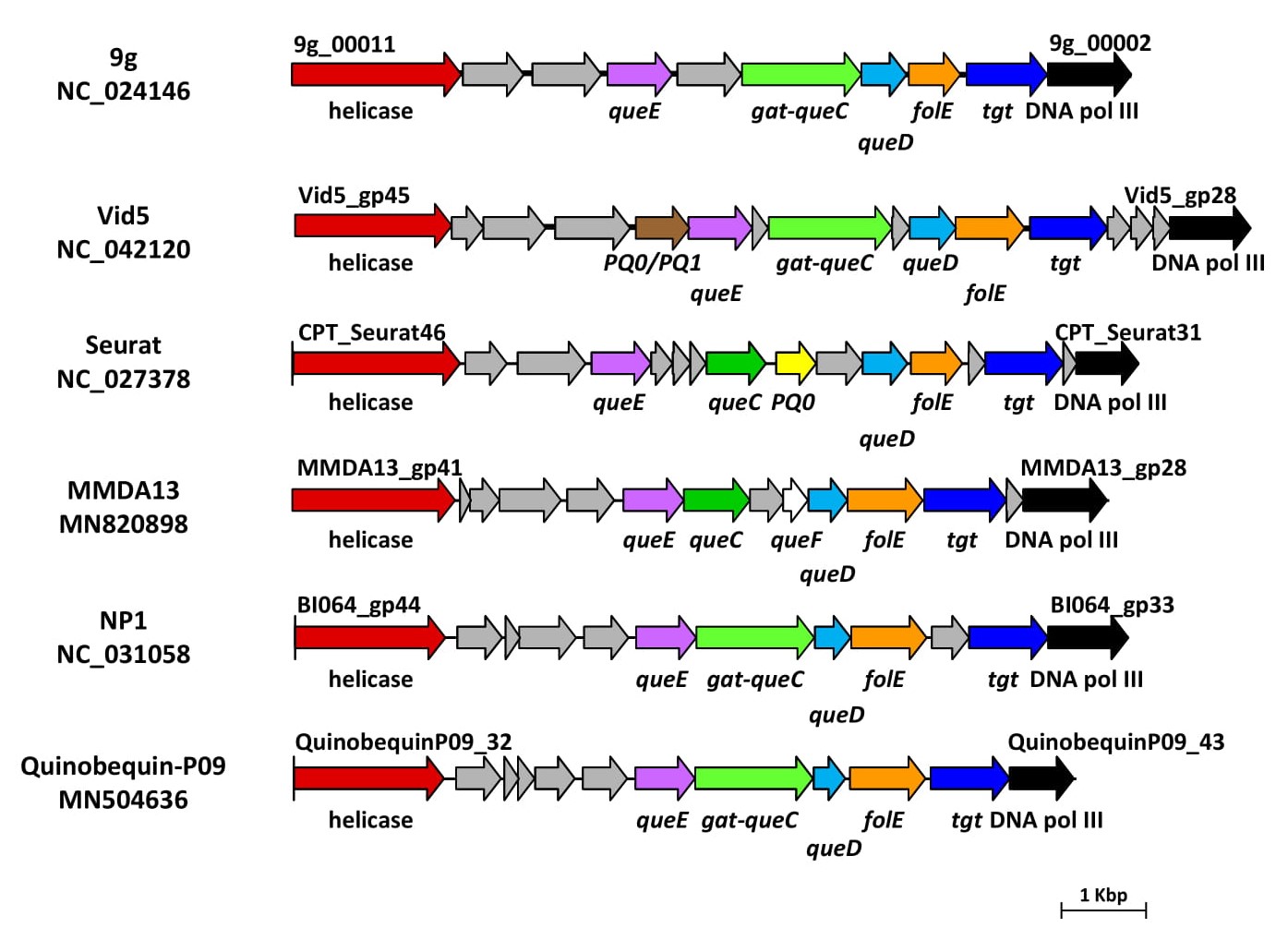
Figure 4. Gene clusters involved in the biosynthesis of 7-deazaguanine derivatives in vB_StuS_MMDA13, in type species of selected International Committee on Taxonomy of Viruses (ICTV) genera, and in the recently characterized Quinobequin-P09 phage. Functions of each deduced protein were assigned by HHpred analysis, by using a ≥90% probability cut-off. Abbreviations are as follows: DNA pol III, DNA polymerase III beta subunit; PQ0/PQ1, PreQ0/PreQ1 transporter; PQ0, PreQ0 transporter. Genes encoding hypothetical proteins are in grey.
Another peculiar feature of vB_StuS_MMDA13 is the host lysis module, formed by five ORFs (gp85-gp89): (i) the endolysin encoding one (gp85); (ii) a holin/antiholin encoding system (gp86) characterized by features typical of a class II holins [11]; (iii) a Rz-like gene (gp87) and (iv) a Rz1-like gene (gp88) which overlaps Rz. The Rz/Rz1 system of vB_StuS_MMDA13, therefore, is of the “overlapped” type [12], and does not share significant similarities with other viral proteins in the databases. The lysis cassette ends with gp89 whose protein product is recognized by HHpred as a peptidoglycan endopeptidase of the RipA, whose members are known to synergistically interact with Rpf(s) in the degradation of peptidoglycan [13].
6. Conclusions
To the best of our knowledge, vB_StuS_MMDA13 is the first characterized lytic phage belonging to Siphoviridae family known to infect members of the Sphingomonas genus.
Indeed, despite the description of several bacteriophages able to lyse members of the Sphingomonadaceae family [14][15][16], very few provide an in depth characterization [3][4][17]. Our results suggest that vB_StuS_MMDA13 doesn't belong to any known bacteriophage genus. In fact, according to Adriaenssens and colleagues [18], a cutoff equal to 40% of shared proteins is the breakpoint to group two Siphoviridae in the same genus, provided the consistence of other features such as genome size and organization, morphology, packaging and replication mechanism. In the case of vB_StuS_MMDA13, the inclusion in one of the described genera should be only possible for the Nipunavirus genus. There are, however, several differences between members of the Nipunavirus and vB_StuS_MMDA13: i) the mean genome size of Nipunavirus (58.2 Kb) is ≈5.5 Kbp smaller than that of our phage (63.7 Kb), so as the ORFs number (74 vs 89) and the GC content (mean of 58.5% for Nipunavirus vs 59.3% of vB_StuS_MMDA13); ii) Nipunavirus infect Pseudomonas aeruginosa, while the host range of vB_StuS_MMDA13 seems restricted to S. turrisvirgatae; iii) the 7-deazaguanidine derivatives biosynthetic pathway is different, with vB_StuS_MMDA13 that lacks a GATase/QueC homologue, and has instead a QueC whose best BLASTP score is with a putative queuosine biosynthesis protein (AGC35898) of the Myoviridae Rhizobium phage RHEph06. The amidotransferase function should be provided by the putative QueF-L which lacks in Nipunavirus.
Moreover, the whole host-lysis module is completely different: it shares no homologies, includes five components in vB_StuS_MMDA13 and four in Nipunavirus, and the putative holins belong to different classes. Finally, some important features of Nipunavirus are not present in vB_StuS_MMDA13, e.g. thymidylate synthase, DNA topoisomerase, and the ribonucleotide reductase of class II. We believe therefore that vB_StuS_MMDA13 should not be regarded as a distant species in the Nipunavirus genus, but should rather represents the type strain of a newly discovered genus, which we propose to be named Ememdadecimater-like virus, after the short name of the first characterized phage of the genus.
References
- Thaller, M.C.; D’Andrea, M.M.; Marmo, P.; Civitareale, C.; Casu, F.; Migliore, L. Sphingomonas turrisvirgatae sp. nov., an agar-degrading species isolated from freshwater. Int. J. Syst. Evol. Microbiol. 2018, 68, 2794–2799.
- Ackermann, H.W.; Eisenstark, A. The present state of phage taxonomy. Intervirology 1974, 3, 201–219.
- Lu, L.; Cai, L.; Jiao, N.; Zhang, R. Isolation and characterization of the first phage infecting ecologically important marine bacteria Erythrobacter. Virol. J. 2017, 14, 104.
- Nielsen, T.K.; Carstens, A.B.; Browne, P.; Lametsch, R.; Neve, H.; Kot, W.; Hansen, L.H. The first characterized phage against a member of the ecologically important sphingomonads reveals high dissimilarity against all other known phages. Sci. Rep. 2017, 7, 13566.
- Yang, Y.; Cai, L.; Ma, R.; Xu, Y.; Tong, Y.; Huang, Y.; Jiao, N.; Zhang, R. A Novel Roseosiphophage Isolated from the Oligotrophic South China Sea. Viruses 2017, 9, 109.
- Flores, V.; Sepúlveda-Robles, O.; Cazares, A.; Kameyama, L.; Guarneros, G. Comparative genomic analysis of Pseudomonas aeruginosa phage PaMx25 reveals a novel siphovirus group related to phages infecting hosts of different taxonomic classes. Arch. Virol. 2017, 162, 2345–2355.
- Hutinet, G.; Kot, W.; Cui, L.; Hillebrand, R.; Balamkundu, S.; Gnanakalai, S.; Neelakandan, R.; Carstens, A.B.; Fa Lui, C.; Tremblay, D.; et al. 7-Deazaguanine modifications protect phage DNA from host restriction systems. Nat. Commun. 2019, 10, 5442.
- Kulikov, E.E.; Golomidova, A.K.; Letarova, M.A.; Kostryukova, E.S.; Zelenin, A.S.; Prokhorov, N.S.; Letarov, A.V. Genomic Sequencing and Biological Characteristics of a Novel Escherichia coli Bacteriophage 9g, a Putative Representative of a New Siphoviridae Genus. Viruses 2014, 6, 5077–5092.
- Šimoliu¯ nas, E.; Šimoliu¯ niene˙ , M.; Kaliniene, L.; Zajancˇkauskaite˙ , A.; Skapas, M.; Meškys, R.; Kaupinis, A.; Valius, M.; Truncaite˙ , L. Pantoea Bacteriophage vB_PagS_Vid5: A Low-Temperature Siphovirus That Harbors a Cluster of Genes Involved in the Biosynthesis of Archaeosine. Viruses 2018, 10, 583.
- Thiaville, J.J.; Kellner, S.M.; Yuan, Y.; Hutinet, G.; Thiaville, P.C.; Jumpathong, W.; Mohapatra, S.; Brochier-Armanet, C.; Letarov, A.V.; Hillebrand, R.; et al. Novel genomic island modifies DNA with 7-deazaguanine derivatives. Proc. Natl. Acad. Sci. USA 2016, 113, E1452–E1459.
- Young, R. Bacteriophage holins: deadly diversity. J. Mol. Microbiol. Biotechnol. 2002, 4, 21–36.
- Summer, E.J.; Berry, J.; Tran, T.A.; Niu, L.; Struck, D.K.; Young, R. Rz/Rz1 Lysis Gene Equivalents in Phages of Gram-negative Hosts. J. Mol. Biol. 2007, 373, 1098–1112.
- Hett, E.C.; Chao, M.C.; Steyn, A.J.; Fortune, S.M.; Deng, L.L.; Rubin, E.J. A partner for the resuscitation-promoting factors of Mycobacterium tuberculosis. Mol. Microbiol. 2007, 66, 658–668.
- Jost, G.; Wiese, J. Temporal variations in the concentrations of bacteria and their lytic phages: an example of an indigenous phage host system in Lake Plußsee, Germany. Fundam. Appl. Limnol. 2013, 182, 183–190.
- Nakayama, N.; Tsuge, T.; Asakawa, S.; Kimura, M. Morphology, host range and phylogenetic diversity of Sphingomonas phages in the floodwater of a Japanese paddy field. Soil Sci. Plant Nutr. 2009, 55, 53–64.
- Wolf, A.; Wiese, J.; Jost, G.; Witzel, K.P. Wide Geographic Distribution of Bacteriophages That Lyse the Same Indigenous Freshwater Isolate (Sphingomonas sp. Strain B18). Appl. Environ. Microbiol. 2003, 69, 2395–2398.
- Zheng, Q.; Chen, Q.; Xu, Y.; Suttle, C.A.; Jiao, N. A Virus Infecting Marine Photoheterotrophic Alphaproteobacteria (Citromicrobium spp.) Defines a New Lineage of ssdna Viruses. Front. Microbiol. 2018, 9, 1418.
- Adriaenssens, E.M.; Edwards, R.; Nash, J.H.E.; Mahadevan, P.; Seto, D.; Ackermann, H.W.; Lavigne, R.; Kropinski, A.M. Integration of genomic and proteomic analyses in the classification of the Siphoviridae family. Virology 2015, 477, 144–154.

