Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Giacomo Casati | + 3042 word(s) | 3042 | 2021-12-27 10:50:11 | | | |
| 2 | Camila Xu | Meta information modification | 3042 | 2022-01-11 02:28:10 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Casati, G. Hippo Pathway in Glioblastoma. Encyclopedia. Available online: https://encyclopedia.pub/entry/17987 (accessed on 13 June 2026).
Casati G. Hippo Pathway in Glioblastoma. Encyclopedia. Available at: https://encyclopedia.pub/entry/17987. Accessed June 13, 2026.
Casati, Giacomo. "Hippo Pathway in Glioblastoma" Encyclopedia, https://encyclopedia.pub/entry/17987 (accessed June 13, 2026).
Casati, G. (2022, January 10). Hippo Pathway in Glioblastoma. In Encyclopedia. https://encyclopedia.pub/entry/17987
Casati, Giacomo. "Hippo Pathway in Glioblastoma." Encyclopedia. Web. 10 January, 2022.
Copy Citation
Glioblastoma (GBM) represents the most common and malignant tumor of the Central Nervous System (CNS), affecting both children and adults. GBM is one of the deadliest tumor types and it shows a strong multidrug resistance (MDR) and an immunosuppressive microenvironment which remain a great challenge to therapy.
glioblastoma (GBM)
signaling pathways
tumor heterogeneity
1. Glioblastoma Chemoresistance
Among the different malignant gliomas, glioblastoma (GBM), which accounts for about 60–70% of all gliomas, is classified as a World Health Organization (WHO) grade IV tumor based on histopathological features, and it represents the most frequent and malignant tumor of the Central Nervous System (CNS), affecting both children and adults with a slight predominance in males.
Despite experimental investigation in this field and the improved therapeutic strategies, GBM remains essentially incurable, with an overall survival time ranging from 12 to 18 months, as less than 5% of patients survive longer than five years after diagnosis. Currently, consolidated first line treatment options for human GBM are radiotherapy and chemotherapy with Temozolomide (TMZ). One hope for a better clinical outcome is to identify targets that play essential roles in mediating the microenvironment-derived survival signal, drug resistance or sensitize the response of GBM cells to radiation and chemotherapeutic drugs. Multidrug resistance (MDR) remains a great challenge to GBM therapy. This cellular phenomenon is the main cause of disease relapse, normal tissue infiltration and distant metastasis.
MDR is due to various and complex mechanisms including crosstalk between tumor microenvironment (TME) and GBM stem cells (GSCs), deregulated signaling pathways, abnormal expression of a specific protein, and cell-to-cell communication mechanisms [1].
The deregulation of the Hippo pathway represents a mechanism that causes MDR in GBM cell lines. In particular, the overexpression of TAZ (Transcriptional Co-activator with PDZ-binding Motif) decreases the cytotoxic effect of TMZ by upregulating the MCL-1 protein and thus making the U87MG and U251 cell lines apoptosis resistant [2].
Moreover, the overexpression of the YAP-TAZ-TEAD complex provokes GBM cell resistance to TMZ treatment by up-regulation of the Hippo pathway downstream target genes. In detail, CTGF (connective tissue growth factor) and Cyr61 (cysteine rich angiogenic inducer 61) genes are upregulated through TGF-β1-dependent activation of Smad/ERK signaling [3].
Moreover, another issue of GBM chemoresistance is due to CD109 protein. CD109 binds to the GP130 receptor and promotes the activation of the IL-6/STAT3 signaling by increasing GSCs stemness and tumorigenicity, and MDR. CD109 can also activate the Hippo pathway in response to damage by conferring radioresistance and chemoresistance through the upregulation of its target genes. Interestingly, the loss of CD109 in vivo leads to a reduction in nuclear YAP (Yes-Associated Protein) level, STAT3 activation and GSCs stemness and therefore reduces tumorigenicity [4][5].
In addition to the intrinsic tumor chemoresistance, drugs delivery through the blood brain barrier (BBB) is very complicated due to abnormal/de novo expression of specific drugs transporters in cancer cells and/or in the endothelial cells forming the BBB [6][7]. Therefore, GBM is characterized by an overexpression of specific drug efflux pumps called ATP-binding cassette (ABC) superfamily such as P-gp, breast cancer resistance protein (BCRP/ABCG2) and MRP1 [8][9]. Furthermore, multiple genetic mechanisms appear to be involved in the resistance phenotype of GBM. GSCs are characterized by altered DNA repair mechanisms, as well DNA damage response (DDR) and mismatch repair (MMR), physiologically involved in the maintenance of genetic stability [10][11][12].
DDR contributes to remove DNA lesions caused by conventional DNA-damaging agent used for GBM such as TMZ and ionizing radiation (IR) conferring chemoresistance phenotype to GBM cells [13]. Failures in MMR are associated with glioma cells TMZ resistance. Repeated exposures to TMZ can induce acquired MSH6 mutations in GBM cells turning off the MSH2/MSH6 dimer and prompting cytotoxicity [14][15].
When the Hippo pathway is “on”, it exploits the DNA repair mechanisms both in immortalized cell lines and in primary cells from GBM patients [16][17]. An intriguing study demonstrated that YAP plays a radioresistant role on gliomas by repairing DNA damage.
After radiation, YAP is able to promote the expression of the FGF2 factor and activate the MAPK-ERK pathway. The YAP-FGF2-MAPK signaling is a key mechanism of radioresistance in GBM [18].
Another study, through a microarray analysis, evaluated the variation of the gene expression profile after proton irradiation. The data show that the Hippo pathway is one of the highly deregulated signaling after proton-therapy irradiation. The Wnt pathway is also deregulated after irradiation. Many authors have already proved that these two molecular signaling interact with each other; the main novelty is that YAP is capable of activating the Wnt/β-catenin pathway, which in turn promotes tumor growth and resistance to radiation in GBM cell lines [19][20][21][22].
The cross-talk between mTOR and Hippo pathway is further evidence of chemoresistance. Indeed, mTORC2 subunit is able to phosphorylate YAP on serine 436 (Ser436) allowing the activation of the Hippo signaling independently from the canonical pathway. This aberrant interplay promotes growth, migration, and drug resistance in both cell lines and GBM patient samples [23][24][25].
About 30–60% of GBM presents methylation of MGMT promoter associated with an increased sensitivity to TMZ and prolonged survival [26][27]. The lack of methylation causes a different correction of the DNA lesion induced by TMZ, generating an incorrect mispairing and consequently an anomalous activation of MMR system. The abnormal loop mode activation of MMR, called “futile cycle”, can provoke the induction of DNA double strand breaks (DSBs) and the activation of specific signaling pathways regarding cellular cycle arrest and cell death. Another molecular mechanism that plays an important role in the genesis and development of chemoresistance in GBM is the aberrant expression of microRNA (miRNAs) [28][29][30][31][32]. miRNAs are short non-coding RNAs molecules that control the expression of genes involved in different cellular processes (proliferation, apoptosis, cell differentiation, anti-viral defense), and their aberrant expression has been reported in tumors [33][34][35][36].
GBM miRNA targets are drug transporter genes, proteins involved in ABCB1/P-gp-mediated chemoresistance and genes involved in DNA repair mechanisms [37][38][39][40][41][42][43][44]. Recent studies, most regarding the release of exosomes, show new interesting data regarding cell-to-cell communication.
Exosomes are cell–cell communication extracellular vesicles with a heterogeneous content of molecules such as protein (receptor, enzymes, and transcription factors), nucleic acids (DNA, mRNA, miRNA, long non-coding RNA (lncRNAs)), growth factors, and lipids [45]. Exosomes participate in both physiological (coagulation and immunosurveillance) and pathological processes (chemoresistance and carcinogenesis) [46][47][48].
Chemoresistance induced by the release of exosomes can involve many cellular pathways such as a TME modulation that induces the epithelium–mesenchymal transition process (EMT). Alternatively, exosome release may activate miRNA-mediated gene expression regulatory mechanisms; furthermore, it may also promote immune escape, angiogenesis and metastasis [49]. Moreover, chemotherapeutic agents can be internalized in exosomes and therefore excluded from drug-resistant tumor cells improving drug efficacy [50]. The delivery of exosomal cargo, which contains drug efflux pumps, fusion genes and lncRNAs, to cancerous cells is associated with drug resistance in GBM [51][52][53][54][55][56].
Another mechanism that contributes to chemoresistance is the inhibition of chemotherapy-induced apoptosis by the tumor. In this regard, the Notch signaling is known to modulate apoptosis in cancer. Specifically, blocking Notch pathway causes the induction of apoptosis. In GBM, inhibition of Notch signaling induces apoptosis in TMZ and Etoposide resistant cells [57][58][59].
However, the Hippo pathway is capable of activating the Notch signaling by the upregulation of JAG-1 protein, thus decreasing apoptosis induced by chemotherapy and increasing chemoresistance [60].
2. Pathways Involved in Glioblastoma Genesis
The GBM development is characterized by many mutations among different key signaling pathways, including the receptor tyrosine kinase (RKT) ones [61][62], such as the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mTOR pathway and the Ras/MAPK/ERK pathway, which are involved in the regulation of cell proliferation, survival, differentiation and angiogenesis. The main tyrosine kinase receptor EGFR (epithelial growth factor receptor) mutation is the EGFR variant III. This alteration maintains the receptor into a constitutionally active ligand-independent form, leading to cell proliferation and survival [63]. EGFR-amplified/mutant human GBMs express a high amount of YAP and VP, an inhibitor of the YAP-TEAD complex, is capable to induce apoptosis in patient-derived EGFR- /mutant GBM because it can suppress expression of YAP/TAZ transcriptional targets, including EGFR. YAP/TAZ-TEAD directly regulates transcription of EGFR itself to create a feedforward loop to drive survival and proliferation of human GBM [64].
EGFR signal transduction also stimulates the Ras/MAPK/ERK pathway resulting in migration and cellular proliferation [65]. It is proven a correlation between receptor tyrosine kinase (RTK) signaling and the Hippo Pathway. Indeed, RTK/RAS driven carcinomas, characterized by chemoresistance, metastasis and tumor invasion, show a dysregulation of the YAP-TEAD complex belonging to the Hippo Pathway. TEAD factor is identified as a migration driver both in vitro and in vivo and as a direct transcriptional target of EGFR. Treatment with VP, not only inhibits cell growth and migration but also causes a dose-dependent downregulation of EGFR activity and ERK phosphorylation [66]. EGFR signal transduction drives the recruitment of PI3K to cell membrane with consequent formation of PIP3 (PI-3-phosphate). PIP3 activates downstream molecules like AKT and mTOR [67]. mTOR and the Hippo Pathway coordinately control cell growth and proliferation. The dysregulation of these signaling plays a critical part in gliomagenesis. It is established a cross-talk between these mechanisms; recent studies consider the Angiomotin protein family as a powerful repressor of YAP [68][69]. In particular, the AMOTL2 protein (angiomotin like-2) is identified as a substrate of the mTORC2 subunit. Indeed in GBM cells, AMOTL2 is phosphorylated at the level of serine 760 by mTORC2. AMOTL2 mutation, that mimics the constitutive phosphorylation of Ser760, stops its ability to bind and suppresses YAP causing a nuclear increase and therefore a greater expression of its oncogenic targets.
Conversely, AMOTL2 overexpression inhibits YAP-induced transcription in vitro [24]. Mutations in the retinoblastoma (RB) pathway are also found in 78% of GBM [70]. RB suppresses cell cycle entry and progression, interacting with the transcription factor E2F [71][72][73]. Other genetic alterations in GBM are at the expense of p53 pathway (altered in 87% of GBM) [70] which is involved in the activation of genes implicated in cell arrest and apoptosis [74]. Hippo Pathway and wild-type p53 cooperate, at multiple levels, as tumor suppressors to cause senescence and apoptosis in response to stressful conditions [75]. Recent in vitro studies show that, in presence of DNA lesions, YAP can interact with p73 (a member of the p53 family) through an independent mechanism of the canonical pathway, inducing apoptosis and reduced proliferation [76][77][78]. In the presence of a mutation, p53 (mtp53) performs an oncogenic activity enhanced by intermediate factors that affect the Hippo pathway. In GBM cells, mtp53 improves PI3K/AKT-mediated phosphorylation of the interacting protein WASP (WIP), a protein associated with the actin cytoskeleton, which promotes YAP stability and cancer stem cell survival [79]. Therefore, Hippo signaling is in the spotlight due to its meaningful roles in both developmental and cancer biology, but it is not the only pathway involved in cell growth and proliferation. These mechanisms are also controlled by other well-known signaling pathways, such as Wnt/β-catenin and TGFβ signaling [80][81].
Indeed, the PI3K/AKT/mTOR pathway is also activated by the Transforming growth factor beta (TGF-β) binding its receptor and in normal conditions it acts as tumor suppressor, inhibiting cell proliferation [82]. Its dysregulation contributes to the GBM pathogenesis, as mutations on TGF-β signaling lead to inflammation, invasion, metastasis, angiogenesis and immune escape [83]. It is demonstrated that various upstream regulators, such as cell polarity, adhesion proteins control Hippo signaling; furthermore, this pathway interacts with other signaling as well Wnt/β-catenin, Notch and MAPK pathways [84]. The role of Wnt pathway in many tumors development, such as in gliomas is well established by several data.
The Wnt pathway contribution in GBM pathology is related to stem cell maintenance and differentiation, tumor initiation and growth, invasion potential and therapeutic resistance, thereby its dysregulation plays an important role in GBM biology [85][86]. In GBM, alterations among this pathway are more frequently found being epigenetic rather than genetic mutations in its signaling components, such as epigenetic silencing of negative Wnt regulators and overexpression of positive ones [87]. Binda et al. underlined the role of Wnt5a (a noncanonical Wnt family member) in brain invasion. The group found that the most invasive gliomas are characterized by Wnt5a overexpression associated with tumor-promoting stem-like characteristics (TPC); indeed, inhibition of Wnt5a in mesenchymal GBM TPC suppresses their infiltration capacity [88]. A cross-talk relationship exists between the Wnt pathway and other important cell signaling pathways such as Notch, Hedgehog, EGFR signaling cascades [89] and the Hippo signaling [90]. Regarding the Notch signaling, it should be underlined its high activation in GSCs (Glioma Stem Cell), where it represses differentiation and preserves stem-like properties, contributing to GBM tumorigenesis and resistance to conventional treatments [91]. Abnormal expression of many Notch components is present in brain tumors. For example, a higher expression of ASCL1, Dll1, Notch 1-3-4, and Hey1, which correlates with higher glioma grade e worse prognosis [92][93]. In addition, Notch signaling activity is reported in WHO grade IV gliomas, and can be associated with hypoxia, PI3K/AKT/mTOR and ERK/MAPK molecular pathway and finally increase malignant features of gliomas [94] (Figure 1).
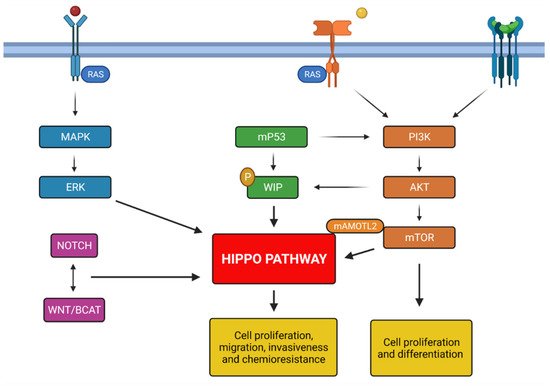
Figure 1. Summary of principals molecular mechanisms involved in glioblastoma genesis; all pathways described activate Hippo pathway promoting the chemoresistance and tumorigenesis. Created with Biorender.com.
3. Background Hippo Pathway
A fine balance between creation of new cells (proliferation) and death of extra ones (apoptosis) is vital to the correct development of organs in all multicellular organisms [95]. Malfunction of these processes contributes to cancer development. One of the main pathways which maintains homeostasis in tissues is the Hippo pathway regulating the appropriate cell number via restriction of cell growth and proliferation, and promoting apoptosis in organ growth [96][97][98]. Therefore, dysregulation of this pathway is a key maker of tumorigenesis and cancer progression [99]. The Hippo pathway’s role was first discovered in Drosophila melanogaster through mosaic genetic screens, whereby were identified several genes that are essential for the appropriate development of adult structures. In 1995 the first identified gene has been warts, encoding the Warts kinase (Wts) [100][101], followed by Salvador, encoding the adaptor protein Salvador (Sav) [102][103] and then, in 2003, Hippo, encoding the Hippo kinase (Hpo) [104][105][106][107]. Mutations on these genes lead to remarkable overgrowth of organ structures in flies, due to the hyperproliferative behavior of these mutated cells which are not pruned away by apoptosis, because of their resistance to it. The core of the Hippo pathway in Drosophila consists of these aforementioned three serine/threonine-protein kinases: Hpo, Sav and Wts. The signal kinase cascade starts with the phosphorylation of Sav by Hpo (in its active form when phosphorylated).
Their interaction leads them to a complex formation that, in turn, phosphorylates Wts and Mats (Mob as-tumor-suppressor). The Salvador kinase is called “adaptor protein”, as it brings Hpo to phosphorylate Wts. Finally, Wst-Mats phosphorylated kinase complex has as its major substrate Yorkie (Yki). As a result of this signaling cascade, Yorkie is inactivated and can not shuttle from the cytoplasm into the nucleus [108]. Homologous of the Hippo-pathway core components have been found in mammals: the mammalian Ste20-like 1 and 2 kinases (MST 1/2 in mammals and Hpo in Drosophila) bind the adaptor SAV1 (the WW-domain containing scaffold protein Salvador; Sav in D.) phosphorylating LATS1/2 (Large Tumor Suppressor homolog 1/2) and MOB1A/B (Mps One Binder 1 cofactor). LATS1/2-MOB1 phosphorylated complex (Wst-Mats complex in D.), in turn, phosphorylates transcriptional downstream co-activators as YAP and TAZ (Yorkie in D.). The kinases cascade regulates the localization of YAP and TAZ, encoded by paralogous genes. In the concrete, by phosphorylation on YAP S127 and on TAZ S89, YAP/TAZ results inactivated as it is forced to remain in the cytoplasm and bind 14-3-3 protein, which will guide YAP/TAZ to a degradation destiny [109]. This is what happens when the Hippo pathway is “on”. Conversely, when the Hippo signaling pathway is “off ”, YAP/TAZ is active as it is able to translocate into the nucleus, where it promotes the transcription of growth promoting or apoptosis inhibition genes. YAP/TAZ transcription co-activator has not DNA binding domains, but it has a TEAD-binding region (TB) and WW domains, characterized by two conserved tryptophan (W) residues. The TB makes it possible for YAP/TAZ to form complexes with TEAD1-4 (transcriptional enhancer factors), while the WW domains allow the interaction of YAP/TAZ with Runx transcription factors, which are involved in carcinogenesis and cancer metastasis [110][111].
Therefore, YAP/TAZ works as a transcription co-activator inducing the expression of target genes such as CTGF, Cyr61, MYC, PD-L1 and FGF-1 (fibroblast growth factor) [112]. Accordingly, a reduction of YAP/TAZ nuclear levels leads to down-regulation of Hippo pathway downstream gene targets. Interestingly, in response to DNA lesions the YAP WW domain interacts with p73 (a p53 family member), resulting in p73 enhanced transcription activity, that induces programmed cell death through transcription of pro-apoptotic genes [113]. Besides the Hippo kinase cascade, multiple signaling pathways and inputs could regulate YAP/TAZ, including Wnt signaling and G-protein coupled receptors (growth-factor signaling pathways), energy stress, mTOR and autophagy [114].
High levels of glucose and the activation of mevalonate pathway are some of the metabolic cues that can trigger YAP/TAZ, some others such as low glucose condition and glucagon stimulation are able to inactivate it [115][116]. YAP/TAZ can in turn influence the metabolism, allowing cell adaptation to the environment [117]. Finally, various upstream signaling mechanisms are involved in the activation of the Hippo core signaling cascade, such as molecular links with adherens junctions (AJs) and tight junctions (TJs) [118][119]. One molecular player between adherens junctions and the Hippo pathway is Neurofibromatosis type 2 (NF2 in mammals and Merlin in D.), an adaptor protein with the FERM-domain encoded by NF2 tumor suppressor gene. NF2 suppresses the activity of YAP in many different cells by activating the Hippo pathway [118]. Thereby, the mechanical stimuli are important for the regulation of the Hippo pathway and consequently, they influence the gene transcription induced by nuclear YAP. Moreover, the Hippo signaling is also affected by cytoskeletal remodeling due to cell junction components binding to F-actin. F-actin stress fibers are present when cultured cells grow on stiff substrates; their presence is correlated with the nuclear localization of YAP. On the contrary, when cells are cultured on soft substrate there is a cytoplasmatic localization of YAP due to lack of stress fibers. Thus, structural changes in the F-actin cytoskeleton lead to an upstream regulation of YAP localization and activity.
References
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233.
- Tian, T.; Li, A.; Lu, H.; Luo, R.; Zhang, M.; Li, Z. TAZ promotes temozolomide resistance by upregulating MCL-1 in human glioma cells. Biochem. Biophys. Res. Commun. 2015, 463, 638–643.
- Zeng, H.; Yang, Z.; Xu, N.; Liu, B.; Fu, Z.; Lian, C.; Guo, H. Connective tissue growth factor promotes temozolomide resistance in glioblastoma through TGF-β1-dependent activation of Smad/ERK signaling. Cell Death Dis. 2017, 8, e2885.
- Filppu, P.; Ramanathan, J.T.; Granberg, K.J.; Gucciardo, E.; Haapasalo, H.; Lehti, K.; Nykter, M.; Le Joncour, V.; Laakkonen, P. CD109-GP130 interaction drives glioblastoma stem cell plasticity and chemoresistance through STAT3 activity. JCI Insight 2021, 6, e141486.
- Minata, M.; Audia, A.; Shi, J.; Lu, S.; Bernstock, J.; Pavlyukov, M.S.; Das, A.; Kim, S.H.; Shin, Y.J.; Lee, Y.; et al. Phenotypic Plasticity of Invasive Edge Glioma Stem-like Cells in Response to Ionizing Radiation. Cell Rep. 2019, 26, 1893–1905.e7.
- Gomez-Zepeda, D.; Taghi, M.; Scherrmann, J.M.; Decleves, X.; Menet, M.C. ABC Transporters at the Blood–Brain Interfaces, Their Study Models, and Drug Delivery Implications in Gliomas. Pharmaceutics 2020, 12, 20.
- Van Tellingen, O.; Yetkin-Arik, B.; De Gooijer, M.C.C.; Wesseling, P.; Wurdinger, T.; De Vries, H.E.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updat. 2015, 19, 1–12.
- Dréan, A.; Rosenberg, S.; Lejeune, F.X.; Goli, L.; Nadaradjane, A.A.; Guehennec, J.; Schmitt, C.; Verreault, M.; Bielle, F.; Mokhtari, K.; et al. ATP binding cassette (ABC) transporters, Expression and clinical value in glioblastoma. J. Neuro-Oncol. 2018, 138, 479–486.
- Declèves, X.; Amiel, A.; Delattre, J.Y.; Scherrmann, J.M.; Decleves, X.; Amiel, A.; Delattre, J.Y.; Scherrmann, J.M. Role of ABC Transporters in the Chemoresistance of Human Gliomas. Curr. Cancer Drug Targets 2006, 6, 433–445.
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760.
- Atkins, R.J.; Ng, W.; Stylli, S.S.; Hovens, C.M.; Kaye, A.H. Repair mechanisms help glioblastoma resist treatment. J. Clin. Neurosci. 2015, 22, 14–20.
- Erasimus, H.; Gobin, M.; Niclou, S.; Van Dyck, E. DNA repair mechanisms and their clinical impact in glioblastoma. Mutat. Res. Rev. Mutat. Res. 2016, 769, 19–35.
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996.
- Cahill, D.P.; Levine, K.K.; Betensky, R.A.; Codd, P.J.; Romany, C.A.; Reavie, L.B.; Batchelor, T.T.; Futreal, P.A.; Stratton, M.R.; Curry, W.T.; et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin. Cancer Res. 2007, 13, 2038–2045.
- Xie, C.; Sheng, H.; Zhang, N.; Li, S.; Wei, X.; Zheng, X. Association of MSH6 mutation with glioma susceptibility, drug resistance and progression. Mol. Clin. Oncol. 2016, 5, 236–240.
- Liu, Z.; Yee, P.P.; Wei, Y.; Liu, Z.; Kawasawa, Y.I.; Li, W. Differential YAP expression in glioma cells induces cell competition and promotes tumorigenesis. J. Cell Sci. 2019, 132, jcs225714.
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaréa, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014.
- Zhang, Y.; Wang, Y.; Zhou, D.; Wang, K.; Wang, X.; Wang, X.; Jiang, Y.; Zhao, M.; Yu, R.; Zhou, X. Radiation-induced YAP activation confers glioma radioresistance via promoting FGF2 transcription and DNA damage repair. Oncogene 2021, 40, 4580–4591.
- Wang, Y.; Pan, P.; Wang, Z.; Zhang, Y.; Xie, P.; Geng, D.; Jiang, Y.; Yu, R.; Zhou, X. β-catenin-mediated YAP signaling promotes human glioma growth. J. Exp. Clin. Cancer Res. 2017, 36, 1–11.
- Kim, S.; Jho, E.H. Merlin, a regulator of Hippo signaling, regulates Wnt/β-catenin signaling. BMB Rep. 2016, 49, 357–358.
- Dong, Z.; Zhou, L.; Han, N.; Zhang, M.; Lyu, X. Wnt/β-catenin pathway involvement in ionizing radiation-induced invasion of U87 glioblastoma cells. Strahlenther Onkol. 2015, 191, 672–680.
- Cammarata, F.P.; Torrisi, F.; Forte, G.I.; Minafra, L.; Bravatà, V.; Pisciotta, P.; Savoca, G.; Calvaruso, M.; Petringa, G.; Cirrone, G.A.P.; et al. Proton Therapy and Src Family Kinase Inhibitor Combined Treatments on U87 Human Glioblastoma Multiforme Cell Line. Int. J. Mol. Sci. 2019, 20, 4745.
- Wu, S.H.; Bi, J.F.; Cloughesy, T.; Cavenee, W.K.; Mischel, P.S. Emerging function of mTORC2 as a core regulator in glioblastoma, Metabolic reprogramming and drug resistance. Cancer Biol. Med. 2014, 11, 255–263.
- Artinian, N.; Cloninger, C.; Holmes, B.; Benavides-Serrato, A.; Bashir, T.; Gera, J. Phosphorylation of the Hippo Pathway Component AMOTL2 by the mTORC2 Kinase Promotes YAP Signaling, Resulting in Enhanced Glioblastoma Growth and Invasiveness. J. Biol. Chem. 2015, 290, 19387–19401.
- Holmes, B.; Benavides-Serrato, A.; Saunders, J.T.; Kumar, S.; Nishimura, R.N.; Gera, J. mTORC2-mediated direct phosphorylation regulates YAP activity promoting glioblastoma growth and invasive characteristics. Neoplasia 2021, 23, 951–965.
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; De Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. Mgmt gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003.
- Martinez, R.; Schackert, G.; Yaya-Tur, R.; Rojas-Marcos, I.; Herman, J.G.; Esteller, M. Frequent hypermethylation of the DNA repair gene MGMT in long-term survivors of glioblastoma multiforme. J. Neurooncol. 2007, 83, 91–93.
- Wong, S.T.; Zhang, X.Q.; Zhuang, J.T.; Chan, H.L.; Li, C.H.; Leung, G.K. MicroRNA-21 inhibition enhances in vitro chemosensitivity of temozolomide-resistant glioblastoma cells. Anticancer Res. 2012, 32, 2835–2841.
- Giunti, L.; Da Ros, M.; Vinci, S.; Gelmini, S.; Iorio, A.L.; Buccoliero, A.M.; Cardellicchio, S.; Castiglione, F.; Genitori, L.; De Martino, M.; et al. Anti-miR21 oligonucleotide enhances chemosensitivity of t98g cell line to doxorubicin by inducing apoptosis. Am. J. Cancer Res. 2015, 5, 231–242.
- Bai, Y.; Liao, H.; Liu, T.; Zeng, X.; Xiao, F.; Luo, L.; Guo, H.; Guo, L. Mir-296-3p regulates cell growth and multi-drug resistance of human glioblastoma by targeting ether-à-go-go (EAG1). Eur. J. Cancer. 2013, 49, 710–724.
- Qian, Z.; Zhou, S.; Zhou, Z.; Yang, X.; Que, S.; Lan, J.; Qiu, Y.; Lin, Y. Mir-146b-5p suppresses glioblastoma cell resistance to temozolomide through targeting TRAF6. Oncol. Rep. 2017, 38, 2941–2950.
- Tian, T.; Mingyi, M.; Qiu, X.; Qiu, Y. MicroRNA-101 reverses temozolomide resistance by inhibition of GSK3β in glioblastoma. Oncotarget 2016, 7, 79584–79595.
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—MicroRNAs with a role in cancer. Nat. Rev. Cancer. 2006, 6, 259–269.
- Visone, R.; Croce, C.M. miRNAs and cancer. Am. J. Pathol. 2009, 174, 1131–1138.
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A MicroRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261.
- Acunzo, M.; Romano, G.; Wernicke, D.; Croce, C.M. MicroRNA and cancer–A brief overview. Adv. Biol. Regul. 2015, 57, 1–9.
- Munoz, J.L.; Rodriguez-Cruz, V.; Ramkissoon, S.H.; Ligon, K.L.; Greco, S.J.; Rameshwar, P. Temozolomide resistance in glioblastoma occurs by miRNA-9-targeted PTCH1, independent of sonic hedgehog level. Oncotarget 2015, 6, 1190–1201.
- Feng, R.; Dong, L. Knockdown of MicroRNA-127 reverses adriamycin resistance via cell cycle arrest and apoptosis sensitization in adriamycin-resistant human glioma cells. Int. J. Clin. Exp. Pathol. 2015, 8, 6107–6116.
- Blower, P.E.; Chung, J.H.; Verducci, J.S.; Lin, S.; Park, J.K.; Dai, Z.; Liu, C.G.; Schmittgen, T.D.; Reinhold, W.C.; Croce, C.M.; et al. MicroRNAs modulate the chemosensitivity of tumor cells. Mol. Cancer Ther. 2008, 7, 1–9.
- Sui, H.; Fan, Z.Z.; Li, Q. Signal transduction pathways and transcriptional mechanisms of ABCB1/PGP-mediated multiple drug resistance in human cancer cells. J. Int. Med. Res. 2012, 40, 426–435.
- Zhao, L.; Lu, X.; Cao, Y. MicroRNA and Signal Transduction Pathways in Tumor Radiation Response. Cell Signal 2013, 25, 1625–1634.
- Taghi, M.; Toossi, B.; Dolat, E.; Khanbabaei, H.; Zafari, N.; Azimian, H. microRNAs, Potential glioblastoma radiosensitizer by targeting radiation-related molecular pathways. Mutat. Res. 2019, 816–818, 111679.
- Szatkowska, M.; Krupa, R. Regulation of DNA Damage Response and Homologous Recombination Repair by microRNA in Human Cells Exposed to Ionizing Radiation. Cancers 2020, 12, 1838.
- Natarajan, V. Regulation of DNA repair by non-coding miRNAs. Noncoding RNA Res. 2016, 1, 64–68.
- Kalra, H.; Drummen, G.P.C.; Mathivanan, S. Focus on Extracellular Vesicles, Introducing the Next Small Big Thing. Int. J. Mol. Sci. 2016, 17, 170.
- Zocco, D.; Ferruzzi, P.; Cappello, F.; Kuo, W.P.; Fais, S. Extracellular vesicles as shuttles of tumor biomarkers and anti-tumor drugs. Front. Oncol. 2014, 4, 267.
- Antonyak, M.A.; Cerione, R.A. Microvesicles as mediators of intercellular communication in cancer. Methods Mol. Biol. 2014, 1165, 147–173.
- Mashouri, L.; Yousefi, H.; Aref, A.R.; Ahadi, A.M.; Molaei, F.; Alahari, S.K. Exosomes, Composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol. Cancer 2019, 18, 75.
- Namee, N.M.C.; O’Driscoll, L. Extracellular vesicles and anti-cancer drug resistance. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 123–136.
- Safaei, R.; Larson, B.J.; Cheng, T.C.; Gibson, M.A.; Otani, S.; Naerdemann, W.; Howell, S.B. Abnormal lysosomal trafficking and enhanced exosomal export of cisplatin in drug-resistant human ovarian carcinoma cell. Mol. Cancer Ther. 2005, 4, 1595–1604.
- Zeng, A.L.; Yan, W.; Liu, Y.W.; Wang, Z.; Hu, Q.; Nie, E.; Zhou, X.; Li, R.; Wang, X.F.; Jiang, T.; et al. Tumour exosomes from cells harbouring PTPRZ1-MET fusion contribute to a malignant phenotype and temozolomide chemoresistance in glioblastoma. Oncogene 2017, 36, 5369–5381.
- De Los Santos, M.C.; Dragomir, M.P.; Calin, G.A. The role of exosomal long non-coding RNAs in cancer drug resistance. Cancer Drug Resist. 2019, 2, 1178–1192.
- Zhang, Z.; Yin, J.; Lu, C.; Wei, Y.; Zeng, A.; You, Y. Exosomal transfer of long non-coding RNA SBF2-AS1 enhances chemoresistance to temozolomide in glioblastoma. J. Exp. Clin. Cancer Res. 2019, 38, 166.
- Shang, C.; Tang, W.; Pan, C.; Hu, X.; Hong, Y. Long non-coding RNA TUSC7 inhibits temozolomide resistance by targeting miR-10a in glioblastoma. Cancer Chemother. Pharmacol. 2018, 81, 671–678.
- Lu, C.; Wei, Y.; Wang, X.; Zhang, Z.; Yin, J.; Li, W.; Chen, L.; Lyu, X.; Shi, Z.; Yan, W.; et al. DNA-methylation-mediated activating of lncRNA SNHG12 promotes temozolomide resistance in glioblastoma. Mol. Cancer 2020, 19, 28.
- Zeng, H.; Xu, N.; Liu, Y.; Liu, B.; Yang, Z.; Fu, Z.; Lian, C.; Guo, H. Genomic profiling of long non-coding RNA and mRNA expression associated with acquired temozolomide resistance in glioblastoma cells. Int. J. Oncol. 2017, 51, 445–455.
- Tomé, M.; Tchorz, J.; Gassmann, M.; Bettler, B. Constitutive activation of Notch2 signalling confers chemoresistance to neural stem cells via transactivation of fibroblast growth factor receptor-1. Stem Cell Res. 2019, 35, 101390.
- Alafate, W.; Xu, D.; Wu, W.; Xiang, J.; Ma, X.; Xie, W.; Bai, X.; Wang, M.; Wang, J. Loss of PLK2 induces acquired resistance to temozolomide in GBM via activation of notch signaling. J. Exp. Clin. Cancer Res. 2020, 39, 1–14.
- Kumar, V.; Vashishta, M.; Kong, L.; Wu, X.; Lu, J.J.; Guha, C.; Dwarakanath, B.S. The Role of Notch, Hedgehog, and Wnt Signaling Pathways in the Resistance of Tumors to Anticancer Therapies. Front Cell Dev. Biol. 2021, 9, 650772.
- Hao, B.; Chen, X.; Cao, Y. Yes-associated protein 1 promotes the metastasis of U251 glioma cells by upregulating Jagged-1 expression and activating the Notch signal pathway. Exp. Ther. Med. 2018, 16, 1411–1416.
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477.
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110.
- Kuan, C.T.; Wikstrand, C.J.; Bigner, D.D. EGF mutant receptor VIII as a molecular target in cancer therapy. Endocr. Relat. Cancer 2001, 8, 83–96.
- Vigneswaran, K.; Boyd, N.H.; Oh, S.Y.; Lallani, S.; Boucher, A.; Neill, S.G.; Olson, J.J.; Read, R.D. YAP/TAZ Transcriptional Coactivators Create Therapeutic Vulnerability to Verteporfin in EGFR-mutant Glioblastoma. Clin. Cancer Res. 2021, 27, 1553–1569.
- Feldkamp, M.M.; Lala, P.; Lau, N.; Roncari, L.; Guha, A. Expression of activated epidermal growth factor receptors, Rasguanosine triphosphate, and mitogen-activated protein kinase in human glioblastoma multiforme specimens. Neurosurgery 1999, 45, 1442–1453.
- Barrette, A.M.; Tome-Garcia, J.; Zaware, N.; Zhou, M.M.; Birtwistle, M.; Tsankova, N. Verteporfin treatment inhibits GBM growth and migration and informs Hippo/RTK crosstalk. Neuro-Oncol. 2018, 20, vi89.
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501.
- Zhao, B.; Li, L.; Lu, Q.; Wang, L.H.; Liu, C.Y.; Lei, Q.Y.; Guan, K.L. Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev. 2011, 25, 51–63.
- Chan, S.W.; Lim, C.J.; Chong, Y.F.; Pobbati, A.V.; Huang, C.X.; Hong, W.J. Hippo Pathway-independent Restriction of TAZ and YAP by Angiomotin. J. Biol. Chem. 2011, 286, 7018–7026.
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068.
- Nevins, J.R. The Rb/E2F pathway and cancer. Hum. Mol. Genet. 2001, 10, 699703.
- Nakada, M. Aberrant signaling pathways in glioma. Cancers 2011, 3, 3242–3278.
- Munro, S.; Carr, S.M.; La Thangue, N.B. Diversity within the pRb pathway: Is there a code of conduct? Oncogene 2012, 31, 4343–4352.
- Harris, S.L.; Levine, A.J. The p53 pathway, positive and negative feedback loops. Oncogene 2005, 24, 2899–2908.
- Raj, N.; Bam, R. Reciprocal Crosstalk between YAP1/Hippo Pathway and the p53 Family Proteins, Mechanisms and Outcomes in Cancer. Front. Cell Dev. Biol. 2019, 7, 159.
- Hamilton, G.; Yee, K.S.; Scrace, S.; O’Neill, E. ATM regulates a RASSF1A-dependent DNA damage response. Curr. Biol. 2009, 19, 2020–2025.
- Van der Weyden, L.; Papaspyropoulos, A.; Poulogiannis, G.; Rust, A.G.; Rashid, M.; Adams, D.J.; Arends, M.J.; O’Neill, E. Loss of RASSF1A synergizes with deregulated RUNX2 signaling in tumorigenesis. Cancer Res. 2012, 72, 3817–3827.
- Pefani, P.E.; O’Neill, E. Hippo pathway and protection of genome stability in response to DNA damage. FEBS J. 2016, 283, 1392–1403.
- Escoll, M.; Gargini, R.; Cuadrado, A.; Anton, I.M.; Wandosell, F. Mutant p53 oncogenic functions in cancer stem cells are regulated by WIP through YAP/TAZ. Oncogene 2017, 36, 3515–3527.
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205.
- Kim, W.; Kim, M.; Jho, E.H. Wnt/beta-catenin signaling: From plasma membrane to nucleus. Biochem. J. 2013, 450, 9–21.
- Lamouille, S.; Derynck, R. Cell size and invasion in TGF-beta-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J. Cell Biol. 2007, 178, 437–451.
- Han, J.; Alvarez-Breckenridge, C.A.; Wang, Q.E.; Yu, J. TGF-beta signaling and its targeting for glioma treatment. Am. J. Cancer Res. 2015, 5, 945–955.
- Ouyang, T.; Meng, W.; Li, M.; Hong, T.; Zhang, N. Recent Advances of the Hippo/YAP Signaling Pathway in Brain Development and Glioma. Cell. Mol. Neurobiol. 2020, 4, 495–510.
- Tompa, M.; Kalovits, F.; Nagy, A.; Kalman, B. Contribution of the Wnt Pathway to Defining Biology of Glioblastoma. Neuromolecular Med. 2018, 20, 437–451.
- Kahlert, U.D.; Maciaczyk, D.; Doostkam, S.; Orr, B.A.; Simons, B.; Bogiel, T.; Reithmeier, T.; Prinz, M.; Schubert, J.; Niedermann, G.; et al. Activation of canonical WNT/β-catenin signaling enhances in vitro motility of glioblastoma cells by activation of ZEB1 and other activators of epithelial-to-mesenchymal transition. Cancer Lett. 2012, 325, 42–53.
- Lee, Y.; Lee, J.K.; Ahn, S.H.; Lee, J.; Nam, D.H. WNT signaling in glioblastoma and therapeutic opportunities. Lab. Investig. 2016, 96, 137–150.
- Binda, E.; Visioli, A.; Giani, F.; Trivieri, N.; Palumbo, O.; Restelli, S.; Dezi, F.; Mazza, T.; Fusilli, C.; Legnani, F.; et al. Wnt5a Drives an Invasive Phenotype in Human Glioblastoma Stem-like Cells. Cancer Res. 2017, 77, 996–1007.
- Suwala, A.K.; Hanaford, A.; Kahlert, U.D.; Maciaczyk, J. Clipping the wings of glioblastoma, Modulation of Wnt as a novel therapeutic strategy. J. Neuropathol. Exp. Neurol. 2016, 75, 388–396.
- Kim, M.; Jho, E.H. Cross talk between Wnt/β-catenin and Hippo signaling pathways: A brief review. BMB Rep. 2014, 47, 540–545.
- Bazzoni, R.; Bentivegna, A. Role of Notch Signaling Pathway in Glioblastoma Pathogenesis. Cancers 2019, 11, 292.
- Somasundaram, K.; Reddy, S.P.; Vinnakota, K.; Britto, R.; Subbarayan, M.; Nambiar, S.; Hebbar, A.; Samuel, C.; Shetty, M.; Sreepathi, H.K.; et al. Upregulation of ASCL1 and inhibition of Notch signaling pathway characterize progressive astrocytoma. Oncogene 2005, 24, 7073–7083.
- Kanamori, M.; Kawaguchi, T.; Nigro, J.M.; Feuerstein, B.G.; Berger, M.S.; Miele, L.; Pieper, R.O. Contribution of Notch signaling activation to human glioblastoma multiforme. J. Neurosurg. 2007, 106, 417–427.
- Gersey, Z.; Osiason, A.D.; Bloom, L.; Shah, S.; Thompson, J.W.; Bregy, A.; Agarwal, N.; Komotar, R.J. Therapeutic targeting of the notch pathway in glioblastoma multiforme. World Neurosurg. 2019, 131, 252–263.e2.
- Conlon, I.; Raff, M. Size control in animal development. Cell 1999, 96, 235–244.
- Harvey, K.; Tapon, N. The Salvador-Warts-Hippo pathway-an emerging tumour-suppressor network. Nat. Rev. Cancer 2007, 7, 182–191.
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828.
- Chen, Y.; Han, H.; Seo, G.; Vargas, R.E.; Yang, B.; Chuc, K.; Zhao, H.; Wang, W. Systematic analysis of the Hippo pathway organization and oncogenic alteration in evolution. Sci. Rep. 2020, 10, 3173.
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257.
- Justice, R.W.; Zilian, O.; Woods, D.F.; Noll, M.; Bryant, P.J. The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 1995, 9, 534–546.
- Xu, T.; Wang, W.; Zhang, S.; Stewart, R.A.; Yu, W. Identifying tumor suppressors in genetic mosaics: The Drosophila lats gene encodes a putative protein kinase. Development 1995, 121, 1053–1063.
- Kango-Singh, M.; Nolo, R.; Tao, C.; Verstreken, P.; Hiesinger, P.R.; Bellen, H.J.; Halderet, G. Shar-pei mediates cell proliferation arrest during imaginal disc growth in Drosophila. Development 2002, 129, 5719–5730.
- Tapon, N.; Harvey, K.F.; Bell, D.W.; Wahrer, D.C.R.; Schiripo, T.A.; Haber, D.A.; Hariharanet, I.K. Salvador Promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell 2002, 110, 467–478.
- Wu, S.; Huang, J.; Dong, J.; Pan, D. Hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 2003, 114, 445–456.
- Udan, R.S.; Kango-Singh, M.; Nolo, R.; Tao, C.; Halder, G. Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat. Cell Biol. 2003, 5, 914–920.
- Harvey, K.F.; Pfleger, C.M.; Hariharan, I.K. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell 2003, 114, 457–467.
- Pantalacci, S.; Tapon, N.; Leopold, P. The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nat. Cell Biol. 2003, 5, 921–927.
- Huang, J.; Wu, S.; Barrera, J.; Matthews, K.; Pan, D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 2005, 122, 421–434.
- Zhao, B.; Li, L.; Tumaneng, K.; Wang, C.Y.; Guan, K.L. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 2010, 24, 72–85.
- Yagi, R.; Chen, L.F.; Shigesada, K.; Murakami, Y.; Ito, Y. A WW domain-containing yes-associated protein (YAP) is a novel tran-scriptional co-activator. EMBO J. 1999, 18, 2551–2562.
- Zaidi, S.K.; Sullivan, A.J.; Medina, R.; Ito, Y.; Van Wijnen, A.J.; Stein, J.L.; Lian, J.B.; Stein, G.S. Tyrosine phosphorylation controls Runx2-mediated subnuclear targeting of YAP to repress transcription. EMBO J. 2004, 23, 790–799.
- Van Rensburg, H.J.J.; Azad, T.; Ling, M.; Hao, Y.; Snetsinger, B.; Khanal, P.; Minassian, L.M.; Graham, C.H.; Rauh, M.J.; Yang, X. The Hippo Pathway Component TAZ Promotes. Immune Evasion in Human Cancer through PD-L1. Cancer Res. 2018, 78, 1457–1470.
- Strano, S.; Munarriz, E.; Rossi, M.; Castagnoli, L.; Shaul, Y.; Sacchi, A.; Oren, M.; Sudol, M.; Cesareni, G.; Blandino, G. Physical inter-action with Yes-associated protein enhances p73 transcriptional activity. J. Biol. Chem. 2001, 276, 15164–15173.
- Liang, N.; Zhang, C.; Dill, P.; Panasyuk, G.; Pion, D.; Koka, V.; Gallazzini, M.N.; Olson, E.; Lam, H.; Henske, E.P.; et al. Regulation of YAP by mTOR and autophagy reveals a therapeutic target of tuberous sclerosis complex. J. Exp. Med. 2014, 211, 2249–2263.
- Santinon, G.; Pocaterra, A.; Dupont, S. Control of YAP/TAZ Activity by Metabolic and Nutrient Sensing Pathways. Trends Cell Biol. 2016, 26, 289–299.
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366.
- Koo, J.H.; Guan, K.L. Interplay between YAP/TAZ and Metabolism. Cell Metab. 2018, 28, 196–206.
- Heng, B.C.; Zhang, X.; Aubel, D.; Bai, Y.; Li, X.; Wei, Y.; Fussenegger, M.; Deng, X. Role of YAP/TAZ in Cell Lineage Fate Determination and Related Signaling Pathways Front Cell. Dev. Biol. 2020, 8, 735.
- Karaman, R.; Halder, R. Cell Junctions in Hippo Signaling. Cold Spring Harb. Perspect. Biol. 2018, 10, a028753.
- Dupont, S. Role of YAP/TAZ in cell-matrix adhesion-mediated signaling and mechanotransduction. Exp. Cell Res. 2016, 343, 42–53.
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183.
- Wada, K.I.; Itoga, K.; Okano, T.; Yonemura, S.; Sasaki, H. Hippo pathway regulation by cell morphology and stress fibers. Development 2011, 138, 3907–3914.
More
Information
Subjects:
Cell Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
2 times
(View History)
Update Date:
11 Jan 2022
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No