Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Shafia Rahman | + 2070 word(s) | 2070 | 2021-12-13 04:38:17 | | | |
| 2 | Yvaine Wei | Meta information modification | 2070 | 2021-12-28 02:32:51 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Rahman, S. Therapeutic Targets of KRAS in Colorectal Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/17575 (accessed on 30 June 2026).
Rahman S. Therapeutic Targets of KRAS in Colorectal Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/17575. Accessed June 30, 2026.
Rahman, Shafia. "Therapeutic Targets of KRAS in Colorectal Cancer" Encyclopedia, https://encyclopedia.pub/entry/17575 (accessed June 30, 2026).
Rahman, S. (2021, December 27). Therapeutic Targets of KRAS in Colorectal Cancer. In Encyclopedia. https://encyclopedia.pub/entry/17575
Rahman, Shafia. "Therapeutic Targets of KRAS in Colorectal Cancer." Encyclopedia. Web. 27 December, 2021.
Copy Citation
Patients with metastatic colorectal cancer have a 5-year overall survival of less than 10%. Approximately 45% of patients with metastatic colorectal cancer harbor KRAS mutations. These mutations not only carry a predictive role for the absence of response to anti-EGFR therapy, but also have a negative prognostic impact on the overall survival.
colorectal cancer
KRAS mutation
1. Introduction
Colorectal cancer (CRC) is one of the most common cancers, with an estimated 1.5 million new cases and 52,980 deaths reported in 2021 in the United States, of which approximately 104,270 arise from the colon and the remainder from the rectum [1]. Although the mortality related to the CRC has been progressively declining since 1990, it continues to be the third most common cause of cancer death in both men and women, respectively, in the United States [2].
CRC arises through a multistep process involving accumulation of various epigenetic and genetic alterations [3]. The pathogenic mechanisms implicated in 80–85% of all CRC cases include microsatellite instability (MSI), CpG island methylator phenotype (CIMP), and chromosomal instability (CIN). The CIN is the most common pathogenic mechanism involved in the development of CRC. It results in the gain or loss of entire or large portions of chromosome resulting in karyotype changes within the cells. These karyotypic changes coupled with the mutations in the tumor suppressor and oncogenes (APC, KRAS, DCC/SMAD4, and TP53) activate oncogenic pathways critical to the pathogenesis of CRC [4]. Mutations in any of the four mismatch repair genes (hMLH1, hMSH2, hMSH6, and hPMS2) result in the microsatellite instability and leads to the development of the hereditary nonpolyposis colorectal cancer (HNPCC), also known as Lynch syndrome [5]. This genetic disorder is inherited as an autosomal dominant pattern and increases the risk of development of several cancers involving the colon, stomach, prostate, and small intestine [4]. The CpG island methylator phenotype is a unique subgroup of CRC. It is characterized by the epigenetic instability resulting in the hypermethylation of CpG island sites at the promoter regions that sequentially leads to transcriptional inactivation of tumor suppressor genes in CRC [5]. In the majority of the cases, one pathway is the predominant pathogenic pathway; however complex interplay in certain cases could be seen [6]. Apart from the above described pathogenic mechanisms, recent studies have shown the alteration in the various metabolic pathways including glycolysis, pyruvate oxidation, lactate oxidation, mitochondrial activity, and glutamine and cholesterol metabolism are involved in the initiation and progression of CRC [7].
The RAS gene family is mutated in approximately 30% of human cancers, with the KRAS isoform mutations being the major contributor [8][9][10]. In Colon cancers, approximately 45% of the cases carry a KRAS mutation [11][12]. These mutations in CRC are associated with aggressive tumor biology and poor survival. Moreover, the KRAS mutations in CRC lead to resistance to epidermal growth factor receptor (EGFR) directed therapies [13].
2. Physiology of RAS
KRAS is located at 12p12.1 and encodes a 188–amino acid residues [14]. The RAS family of genes acts as a universal confluence in the signal transduction of multiple intracellular pathways. KRAS and other RAS oncogenes are intracellular guanine nucleotide-binding proteins (G proteins) that belong to the family of small GTPases and function as GDP/GTP-regulated molecular switches [15]. It is activated by varying signals ranging from growth factors (epidermal growth factor receptor, platelet-derived growth factor receptor, insulin like growth factor, etc.), hormones, and cytokines to neurotransmitters [14]. Once activated, RAS moves from an inactivated, GDP-bound form, to activated GTP- bound state. The activation is catalyzed by guanine nucleotide exchange-factors (GEFs) and the conversion back to inactivated form by GTP hydrolysis mediated by GTPase-activating proteins (GAPs) [16]. The RAS activates multiple downstream pathways including the RAS-RAF-MEK-ERK pathway, which regulates cell cycle and proliferation [16]. Another pathway involved is PI3K-AKT-mTOR, which also promotes cell growth and suppresses apoptosis. The RAS-related protein (RAL) pathway and the tumor invasion and metastasis-inducing protein 1 (TIAM1-RAC1) are involved in intracellular vesicle trafficking, cytoskeletal organization, and tumor growth [17]. Thus, RAS proteins are essential regulators of the various aspects of normal cell growth and physiology and play a role in malignant transformation (Figure 1). Apart from playing critical role in the signal transduction involving multiple intracellular pathways, oncogenic KRAS is known to dysregulate various metabolic processes including glutaminolysis, glycolysis, and redox hemostasis promoting tumorigenesis and chemoresistance [18].
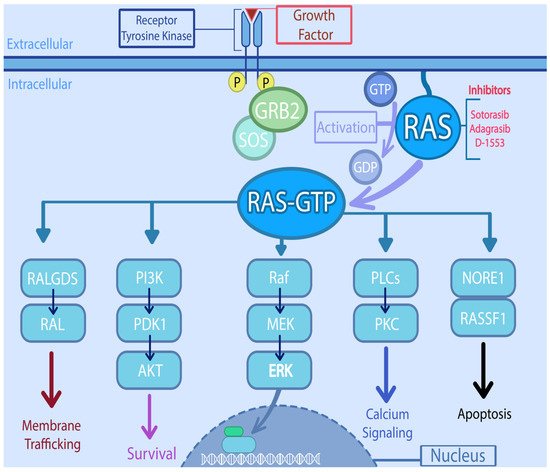
Figure 1. Signaling pathways downstream of RAS and potential targets. RAS directly activates the mitogen activated protein kinase (MAPK) cascade, through phosphorylation of Raf (Rapidly accelerated fibro sarcoma) which in turn phosphorylates MEK (Mitogen activated protein kinase kinase), which then phosphorylates MAPK. On the other hand, it also interacts with the PI3K (Phosphatidylinositol-4,5-Bisphosphate 3-Kinase)/AKT (serine/threonine protein kinase) pathway, either by PI3K interaction or through RAC1 which in turn activates p21-activated kinase (PAK), an AKT interacting protein. RAS also activates the RAL (RAS like proto-oncogene) which is involved in various steps of membrane trafficking. The PLCs (Phospholipase C) along with RINI/ABL plays important role in cytoskeletal remodeling. The activation of NORE1/RASSF1 is involved in cell cycle arrest and apoptosis.
3. Mutations Involving RAS in CRC
RAS mutations have been associated with aberrant cell signaling that leads to tumor-promoting inflammation and play a key role in carcinogenesis by inducing an array of inflammatory cytokines, chemokines and accentuates tumorigenesis and invasiveness. The RAS mutations are common in CRC (~45%), with KRAS being the most prevalent (85%), followed by NRAS (15%) and HRAS (1%) [19]. The majority of the KRAS mutations in the CRC are located in codons 12 and 13 of exon 2 (80% are G12A, G12C, G12D, G12S, G12V, G13C, G13D), and less frequently in codon 61 of exon 3 (5% are Q61H, Q61L, and Q61R) and codon 146 of exon 4 (2% are A146T and A146V) [20]. Mutations in any of these codons promote the accelerated exchange of nucleotides, and a decrease in the binding of GAP. Either of these leads to increase GTP binding and KRAS activation. KRAS mutations also carry a predictive role for the absence of response to anti-EGFR therapy in metastatic CRC and thus have a negative prognostic impact as well [21][22].
4. Targeting KRAS
The therapeutic strategies under investigation to target KRAS mutations in CRC includes therapy directed towards mutant KRAS, targeting KRAS-membrane association, and the combined inhibition of downstream pathways.
4.1. KRAS Directed
Several studies are being performed to identify molecules able to bind the mutated sites of KRAS or inhibit the synthesis at the DNA level of the mutated protein and subsequently blocking the activity of KRAS.
4.2. AMG 510
AMG 510 (Sotorasib) is the first FDA-approved specific, irreversible inhibitor of KRAS G12C. It traps the KRAS in the inactive GDP-bound state [23]. AMG 510 has shown in the preclinical studies to inhibit phosphorylation of extracellular signal-regulated kinase (ERK), a critical downstream effector of KRAS, producing a durable complete tumor regression in mice bearing KRAS p.G12C tumors [24]. Based on the significant objective response rate and duration of response in a phase 1 trial CodeBreak100 (NCT03600883) [25], it was approved in locally advanced or metastatic NSCLC. Although the KRAS G12C is noted only in 1–3% of CRC, the recent promising clinical data breaks the assumption of KRAS being undruggable [26].
4.3. MRTX849
Another direct target of KRAS is MRTX849 (adagrasib). It works by irreversibly and selectively binding to KRAS G12C in its inactive state, blocking its signaling to other cells, thus preventing tumor cell growth and proliferation, leading to cancer cell death [27]. KRYSTAL-1 phase I/II clinical trial showed clinical activity in non-small cell lung cancer (NSCLC), CRC and other solid tumors such as pancreatic, endometrial, and ovarian cancers [28]. The FDA granted breakthrough therapy designation to MRTX849 for the treatment of patients with KRAS G12C-mutated non-small cell lung cancer patients that has previously received systemic therapy.
5. Targeting Membrane Association
Targeting of G4 Structures
G-quadruplex (G4) structures are DNA tetraplexes that typically form in guanine-rich regions of genomes. Four guanine bases bind with Hoogsteen hydrogen bonds to form a guanine tetrad plane (G-quartet), and then two or more G-quartet planes stack on top of each other to form a G4 structure [29] Figure 2. The G4 structures are abundant in the promoter regions of many genes, including the regulation transcription of oncogenes and tumor suppressor genes [30][31][32]. Other than being reported on the KRAS human promoter DNA, G4 structures are found in RNA sequences, including the 5′ untranslated region (UTRs) of KRAS mRNA. Purro et al. identified natural alkaloids Indoloquinolines as potential G4-ligand compounds for targeting of KRAS in CRC [33][34]. They also synthesized a new molecule, namely EMICORON, which binds to the G4 structures on KRAS. Treatment with EMICORON, downregulated KRAS mRNA and protein expression in CRC cell lines, with decreased tumor volume in KRAS-mutated patient-derived xenografts [33]. It was further shown the EMICORON co- administration with FOLFIRI, improved the efficacy of chemotherapy in CRC-bearing mice [33].
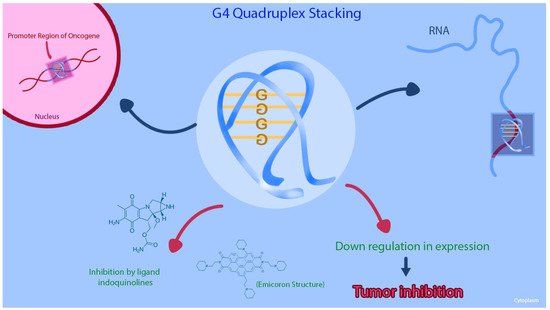
Figure 2. This figure shows that the G-quadruplex (G4) structures and the inhibition of the G4 structure on KRAS promoter region can cause downregulation of gene expression in CRC cell lines.
6. Indirect Approaches
6.1. PDEδ Inhibition
Prenyl-binding protein PDEδ is crucial in maintaining the spatial organization of RAS during the activation of signaling pathway [35][36]. This creates a novel mechanism of indirectly targeting RAS signaling through inhibition of PDEδ. Zimmermann et al., developed Deltarasin, a small molecule inhibitor of PDEδ and demonstrated in the pre-clinical study that PDEδ inhibition by Deltarasin not only blocked the oncogenic RAS signaling but suppressed both in vitro and in vivo proliferation of human pancreatic ductal adenocarcinoma cells with highly prevalent oncogenic KRAS mutant genes [32]. Based on this pre-clinical data various small molecule inhibitors of PDEδ were generated. These inhibitors, such as Deltazinone 1 and Deltasonamide 1 and 2, competitively interact with the farnesyl-binding pocket of RAS [37][38]. Another pre-clinical study supporting the above strategy was done by Klein et al., [39]. They successfully demonstrated the inhibitory effect of PDEδ blockage on the proliferation and survival of KRAS mutant CRC cell lines.
6.2. Targeting NRF2/Oxidative and Glutaminolysis
One of the major cellular changes which drive the proliferation in the cancer cells is the ability to induce metabolic reprogramming [37]. The normal cells induce glycolysis as a source of ATP in response to hypoxia. However, the cancer cells express exorbitant aerobic glycolysis promoting rapid cellular proliferation. This phenomenon is known as the Warburg’s effect [38]. One of the major sources of substrates for anerobic glycolysis is glutamine. Glutamine is an important amino acid necessary for the synthesizing glutamate via glutaminolysis, which in turn contributes to the tricarboxylic acid (TCA) cycle in the absence of glucose [39], thus making the cancer cells dependent on the glutamine-mediated TCA cycle for their energy needs. However, the rapid cellular proliferation and metabolism rate generates oxidative radicals deleterious for cell survival. The nuclear factor-erythroid 2 -related factor 2 (NRF2) and Kelch-like ECH-associated protein 1 (KEAP1) pathway protects the cells against oxidative and electrophilic stress and is tightly regulated under normal physiological conditions [40]. In cancer cells, apart from the enhanced glutaminolysis, dysregulation of NRF2/oxidative, inhibition of repressor genes, pathway promotes constant detoxification and transcription of cytoprotective proteins. Multiple experimental studies have shown that mutant KRAS enhances the activation of NRF2 antioxidant system and gene expression of the enzymes involved in the glutaminolysis, promoting tumorigenesis [41][42]. Given this critical role of glutaminolysis and NRF2/oxidative pathway, it has become an exciting therapeutic target to combat KRAS driven cancers. Furthermore, the glutaminase and NRF2 inhibitors have shown to enhance sensitivity of cancer cells to chemotherapy.
7. Developing Therapies
7.1. MiRNA as Potential Drug Candidates
MicroRNAs (miRNA) are small, approximately 20-nucleotide long, single stranded, non-coding RNA molecules and regulate gene expression by binding to complementary 3′ untranslated region (UTR) of a target gene leading to either degradation of mRNA or inhibition of translation [43]. Multiple studies have elucidated the critical role of miRNAs in cell proliferation, migration, invasion, apoptosis, and angiogenesis [44]. These miRNAs can function as either a tumor suppressor or an oncogene in the regulation pathway, depending on the cell context. For example, an miR-96 is upregulated in lung, prostate, bladder, colorectal, and breast cancer however the same miR-96 is downregulated in pancreatic cancer [45]. Chan et al. experimentally showed that miR-143 downregulation was associated with upregulation of KRAS protein in CRC cell lines. Upon treating CRC cell lines with miR-143, suppression of KRAS protein translation was observed. However, when miR-143 inhibitor was used it stimulated cell proliferation and increased the KRAS protein level [46].
7.2. MYC Inhibition
The RAS and the MYC oncogenes interplay and interdependency play an essential role in driving cancer development [47]. Several studies in mouse models have demonstrated the importance of MYC in KRAS-driven oncogenesis and genetic suppression of MYC impairing the growth of KRAS-driven cancer cells [47][48]. The MYC oncoprotein in KRAS mutant cells is stabilized via increased MYC transcription and decreased protein degradation mediated by CDK 9 directed phosphorylation of MYC [49]. Thus, targeting the MYC oncogene could be a potential therapeutic strategy for MYC-dependent cancers such as KRAS-mutant CRC [50]. Voruciclib is a cyclin-dependent kinase (CDK) inhibitor and selectively inhibits cyclin-dependent kinase 4 (CDK4) and 6 (CDK6) [51]. This, in turn, blocks the phosphorylation of the retinoblastoma protein in early G1 phase, preventing the CDK-mediated G1-S phase transition leading to cell cycle arrest. The suppression of replication of DNA, in turn, inhibits tumor cell proliferation. The anti-neoplastic potential of voruciclib arises from its activity to inhibit the Cyclin-dependent Kinases.
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33.
- Islami, F.; Ward, E.M.; Sung, H.; Cronin, K.A.; Tangka, F.K.L.; Sherman, R.L.; Zhao, J.; Anderson, R.N.; Henley, S.J.; Yabroff, K.R.; et al. Annual Report to the Nation on the Status of Cancer, Part 1: National Cancer Statistics. J. Natl. Cancer Inst. 2021, 113, 1648–1669.
- Loeb, L.A.; Springgate, C.F.; Battula, N. Errors in DNA Replication as a Basis of Malignant Changes. Cancer Res. 1974, 34, 2311–2321.
- Pino, M.S.; Chung, D.C. The Chromosomal Instability Pathway in Colon Cancer. Gastroenterology 2010, 138, 2059–2072.
- Vilar, E.; Gruber, S.B. Microsatellite instability in colorectal cancer—The stable evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162.
- Pancione, M.; Remo, A.; Colantuoni, V. Genetic and Epigenetic Events Generate Multiple Pathways in Colorectal Cancer Progression. Pathol. Res. Int. 2012, 2012, 509348.
- La Vecchia, S.; Sebastián, C. Metabolic pathways regulating colorectal cancer initiation and progression. Semin. Cell Dev. Biol 2020, 98, 63–70.
- Zhang, Z.; Wang, Y.; Vikis, H.G.; Johnson, L.; Liu, G.; Li, J.; Anderson, M.W.; Sills, R.C.; Hong, H.L.; Devereux, T.R.; et al. Wildtype Kras2 can inhibit lung carcinogenesis in mice. Nat. Genet. 2001, 29, 25–33.
- Anderson, M.W.; Reynolds, S.H.; You, M.; Maronpot, R.M. Role of proto-oncogene activation in carcinogenesis. Environ. Health Perspect. 1992, 98, 13–24.
- Kranenburg, O. The KRAS oncogene: Past, present, and future. Biochim. Biophys. Acta 2005, 1756, 81–82.
- Bos, J.L.; Fearon, E.R.; Hamilton, S.R.; Vries, M.V.d.; van Boom, J.H.; van der Eb, A.J.; Vogelstein, B. Prevalence of ras gene mutations in human colorectal cancers. Nature 1987, 327, 293–297.
- Bazan, V.; Agnese, V.; Corsale, S.; Calò, V.; Valerio, M.R.; Latteri, M.A.; Vieni, S.; Grassi, N.; Cicero, G.; Dardanoni, G.; et al. Specific TP53 and/or Ki-ras mutations as independent predictors of clinical outcome in sporadic colorectal adenocarcinomas: Results of a 5-year Gruppo Oncologico dell’Italia Meridionale (GOIM) prospective study. Ann. Oncol. 2005, 16, iv50–iv55.
- Überall, I.; Kolář, Z.; Trojanec, R.; Berkovcová, J.; Hajdúch, M. The status and role of ErbB receptors in human cancer. Exp. Mol. Pathol. 2008, 84, 79–89.
- Jančík, S.; Drábek, J.; Radzioch, D.; Hajdúch, M. Clinical Relevance of KRAS in Human Cancers. J. Biomed. Biotechnol. 2010, 2010, 150960.
- Hancock, J.F.; Magee, A.I.; Childs, J.E.; Marshall, C.J. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell 1989, 57, 1167–1177.
- Seger, R.; Krebs, E.G. The MAPK signaling cascade. FASEB J. 1995, 9, 726–735.
- Lambert, J.M.; Lambert, Q.T.; Reuther, G.W.; Malliri, A.; Siderovski, D.P.; Sondek, J.; Collard, J.G.; Der, C.J. Tiam1 mediates Ras activation of Rac by a PI(3)K-independent mechanism. Nat. Cell Biol. 2002, 4, 621–625.
- Mukhopadhyay, S.; Vander Heiden, M.G.; McCormick, F. The Metabolic Landscape of RAS-Driven Cancers from biology to therapy. Nat. Cancer 2021, 2, 271–283.
- Vaughn, C.P.; Zobell, S.D.; Furtado, L.V.; Baker, C.L.; Samowitz, W.S. Frequency of KRAS, BRAF, and NRAS mutations in colorectal cancer. Genes Chromosomes Cancer 2011, 50, 307–312.
- Li, Z.-N.; Zhao, L.; Yu, L.-F.; Wei, M.-J. BRAF and KRAS mutations in metastatic colorectal cancer: Future perspectives for personalized therapy. Gastroenterol. Rep. 2020, 8, 192–205.
- Zocche, D.M.; Ramirez, C.; Fontao, F.M.; Costa, L.D.; Redal, M.A. Global impact of KRAS mutation patterns in FOLFOX treated metastatic colorectal cancer. Front. Genet. 2015, 6, 116.
- Esteller, M.; González, S.; Risques, R.A.; Marcuello, E.; Mangues, R.; Germà, J.R.; Herman, J.G.; Capellà, G.; Peinado, M.A. K-ras and p16 aberrations confer poor prognosis in human colorectal cancer. J. Clin. Oncol. 2001, 19, 299–304.
- Lito, P.; Solomon, M.; Li, L.-S.; Hansen, R.; Rosen, N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 2016, 351, 604–608.
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223.
- Fakih, M.; Desai, J.; Kuboki, Y.; Strickler, J.H.; Price, T.J.; Durm, G.A.; Falchook, G.S.; Denlinger, C.S.; Krauss, J.C.; Shapiro, G.; et al. CodeBreak 100: Activity of AMG 510, a novel small molecule inhibitor of KRASG12C, in patients with advanced colorectal cancer. J. Clin. Oncol. 2020, 38, 4018.
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217.
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71.
- Sabari, J.K.; Park, H.; Tolcher, A.W.; Ou, S.-H.I.; Garon, E.B.; George, B.; Janne, P.A.; Moody, S.E.; Tan, E.Y.; Sen, S.K.; et al. KRYSTAL-2: A phase I/II trial of adagrasib (MRTX849) in combination with TNO155 in patients with advanced solid tumors with KRAS G12C mutation. J. Clin. Oncol. 2021, 39, TPS146.
- Wu, F.; Niu, K.; Cui, Y.; Li, C.; Lyu, M.; Ren, Y.; Chen, Y.; Deng, H.; Huang, L.; Zheng, S.; et al. Genome-wide analysis of DNA G-quadruplex motifs across 37 species provides insights into G4 evolution. Commun. Biol. 2021, 4, 98.
- Brooks, T.A.; Kendrick, S.; Hurley, L. Making sense of G-quadruplex and i-motif functions in oncogene promoters. FEBS J. 2010, 277, 3459–3469.
- Cheng, Y.; Tang, Q.; Li, Y.; Zhang, Y.; Zhao, C.; Yan, J.; You, H. Folding/unfolding kinetics of G-quadruplexes upstream of the P1 promoter of the human BCL-2 oncogene. J. Biol. Chem. 2019, 294, 5890–5895.
- Lavrado, J.; Borralho, P.M.; Ohnmacht, S.A.; Castro, R.E.; Rodrigues, C.M.; Moreira, R.; dos Santos, D.J.; Neidle, S.; Paulo, A. Synthesis, G-quadruplex stabilisation, docking studies, and effect on cancer cells of indoloquinolines with one, two, or three basic side chains. ChemMedChem 2013, 8, 1648–1661.
- Porru, M.; Artuso, S.; Salvati, E.; Bianco, A.; Franceschin, M.; Diodoro, M.G.; Passeri, D.; Orlandi, A.; Savorani, F.; D’Incalci, M.; et al. Targeting G-Quadruplex DNA Structures by EMICORON Has a Strong Antitumor Efficacy against Advanced Models of Human Colon Cancer. Mol. Cancer 2015, 14, 2541–2551.
- Porru, M.; Zizza, P.; Franceschin, M.; Leonetti, C.; Biroccio, A. EMICORON: A multi-targeting G4 ligand with a promising preclinical profile. Biochim. Biophys. Acta 2017, 1861, 1362–1370.
- Chandra, A.; Grecco, H.E.; Pisupati, V.; Perera, D.; Cassidy, L.; Skoulidis, F.; Ismail, S.A.; Hedberg, C.; Hanzal-Bayer, M.; Venkitaraman, A.R.; et al. Erratum: The GDI-like solubilizing factor PDEδ sustains the spatial organization and signalling of Ras family proteins. Nat. Cell Biol. 2012, 14, 329.
- Zhang, H.; Liu, X.-h.; Zhang, K.; Chen, C.-K.; Frederick, J.M.; Prestwich, G.D.; Baehr, W. Photoreceptor cGMP Phosphodiesterase δ Subunit (PDEδ) Functions as a Prenyl-binding Protein. J. Biol. Chem. 2004, 279, 407–413.
- Papke, B.; Murarka, S.; Vogel, H.A.; Martín-Gago, P.; Kovacevic, M.; Truxius, D.C.; Fansa, E.K.; Ismail, S.; Zimmermann, G.; Heinelt, K.; et al. Identification of pyrazolopyridazinones as PDEδ inhibitors. Nat. Commun. 2016, 7, 11360.
- Martín-Gago, P.; Fansa, E.K.; Klein, C.H.; Murarka, S.; Janning, P.; Schürmann, M.; Metz, M.; Ismail, S.; Schultz-Fademrecht, C.; Baumann, M.; et al. A PDE6δ-KRas Inhibitor Chemotype with up to Seven H-Bonds and Picomolar Affinity that Prevents Efficient Inhibitor Release by Arl2. Angew. Chem. Int. Ed. Engl. 2017, 56, 2423–2428.
- Klein, C.H.; Truxius, D.C.; Vogel, H.A.; Harizanova, J.; Murarka, S.; Martín-Gago, P.; Bastiaens, P.I.H. PDEδ inhibition impedes the proliferation and survival of human colorectal cancer cell lines harboring oncogenic KRas. Int. J. Cancer 2019, 144, 767–776.
- Maitra, R.; Ghalib, M.H.; Goel, S. Reovirus: A targeted therapeutic--progress and potential. Mol. Cancer Res. 2012, 10, 1514–1525.
- Schmitz, K.J.; Ademi, C.; Bertram, S.; Schmid, K.W.; Baba, H.A. Prognostic relevance of autophagy-related markers LC3, p62/sequestosome 1, Beclin-1 and ULK1 in colorectal cancer patients with respect to KRAS mutational status. World J. Surg. Oncol 2016, 14, 189.
- Jiffry, J.; Thavornwatanayong, T.; Rao, D.; Fogel, E.J.; Saytoo, D.; Nahata, R.; Guzik, H.; Chaudhary, I.; Augustine, T.; Goel, S.; et al. Oncolytic Reovirus (pelareorep) Induces Autophagy in KRAS-mutated Colorectal Cancer. Clin. Cancer Res. 2021, 27, 865–876.
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol 2014, 15, 509–524.
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal. Transduct. Target. Ther. 2016, 1, 15004.
- Hong, Y.; Liang, H.; Uzair Ur, R.; Wang, Y.; Zhang, W.; Zhou, Y.; Chen, S.; Yu, M.; Cui, S.; Liu, M.; et al. miR-96 promotes cell proliferation, migration and invasion by targeting PTPN9 in breast cancer. Sci. Rep. 2016, 6, 37421.
- Chen, X.; Guo, X.; Zhang, H.; Xiang, Y.; Chen, J.; Yin, Y.; Cai, X.; Wang, K.; Wang, G.; Ba, Y.; et al. Role of miR-143 targeting KRAS in colorectal tumorigenesis. Oncogene 2009, 28, 1385–1392.
- Soucek, L.; Whitfield, J.R.; Sodir, N.M.; Massó-Vallés, D.; Serrano, E.; Karnezis, A.N.; Swigart, L.B.; Evan, G.I. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 2013, 27, 504–513.
- Soucek, L.; Whitfield, J.; Martins, C.P.; Finch, A.J.; Murphy, D.J.; Sodir, N.M.; Karnezis, A.N.; Swigart, L.B.; Nasi, S.; Evan, G.I. Modelling Myc inhibition as a cancer therapy. Nature 2008, 455, 679–683.
- Blake, D.R.; Vaseva, A.V.; Hodge, R.G.; Kline, M.P.; Gilbert, T.S.K.; Tyagi, V.; Huang, D.; Whiten, G.C.; Larson, J.E.; Wang, X.; et al. Application of a MYC degradation screen identifies sensitivity to CDK9 inhibitors in KRAS-mutant pancreatic cancer. Sci. Signal. 2019, 12.
- Ischenko, I.; Zhi, J.; Hayman, M.J.; Petrenko, O. KRAS-dependent suppression of MYC enhances the sensitivity of cancer cells to cytotoxic agents. Oncotarget 2017, 8, 17995–18009.
- Gupta, P.; Zhang, Y.K.; Zhang, X.Y.; Wang, Y.J.; Lu, K.W.; Hall, T.; Peng, R.; Yang, D.H.; Xie, N.; Chen, Z.S. Voruciclib, a Potent CDK4/6 Inhibitor, Antagonizes ABCB1 and ABCG2-Mediated Multi-Drug Resistance in Cancer Cells. Cell. Physiol. Biochem. 2018, 45, 1515–1528.
More
Information
Subjects:
Oncology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
861
Revisions:
2 times
(View History)
Update Date:
29 Mar 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No