Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Zhang, H. Ferroptosis in NAFLD. Encyclopedia. Available online: https://encyclopedia.pub/entry/17257 (accessed on 09 August 2026).
Zhang H. Ferroptosis in NAFLD. Encyclopedia. Available at: https://encyclopedia.pub/entry/17257. Accessed August 09, 2026.
Zhang, Han. "Ferroptosis in NAFLD" Encyclopedia, https://encyclopedia.pub/entry/17257 (accessed August 09, 2026).
Zhang, H. (2021, December 17). Ferroptosis in NAFLD. In Encyclopedia. https://encyclopedia.pub/entry/17257
Zhang, Han. "Ferroptosis in NAFLD." Encyclopedia. Web. 17 December, 2021.
Copy Citation
Non-alcoholic fatty liver disease (NAFLD) is a chronic progressive liver disease with steatosis as the main pathological feature, including simple fatty liver degeneration, non-alcoholic steatohepatitis (NASH). It may develop into cirrhosis and liver cancer. NAFLD is the most common chronic liver disease in the world today, and its incidence in the Euro-American region has reached more than 20%.
ferroptosis
NAFLD
iron metabolism
Mechanism
1. Ferroptosis and Its Mechanism
The term “ferroptosis” was first proposed by Dixon et al., in 2012, and it was used to describe a non-apoptotic form of cell death caused by erastin-induced iron-dependent lipid peroxide accumulation [1]. The main morphological characteristics of ferroptosis are the shrinkage of cell volume, the decrease of mitochondrial cristae, and the increase of mitochondrial membrane density without typical apoptotic or necrotic manifestation [1]. The main biochemical changes of ferroptosis are the depletion of glutathione (γ-glutamylcysteinylglycine, GSH) and decrease in glutathione peroxidase 4 (GPX4) activity [1]. Subsequently, the divalent iron ions oxidize lipids in a manner similar to the Fenton reaction, thereby generating a large number of reactive oxygen species (ROS), which in turn promote ferroptosis of cells [2]. GPX4 is the main enzyme that protects the cell membrane against peroxidative damage [3][4], and either its direct or indirect inactivation or an increase in the unstable iron pool will induce the occurrence of ferroptosis [1]. In addition, iron chelator (deferoxamine) and some small molecule compounds (such as ferrostatin-1 (Fer-1) and lipoxstatin-1 (Lip-1)) can reverse the lipid peroxidation caused by ferroptosis [1][2]. It has also been found that nitric oxide synthase (iNOS) may also act as a ferroptosis inhibitor by scavenging lipid free radicals or free radical intermediates [5].
Generally, ferroptosis has a complicated network of genes, proteins, and metabolism. Abnormal iron metabolism and lipid peroxidation may be the main factors that cause ferroptosis, and the system Xc−/GSH/GPX4 axis plays a crucial role in this process. In addition, ferroptosis suppressor protein (FSP1), nuclear factor erythroid 2-related factor 2 (Nrf2), p53, and DHODH also have important regulatory effects on ferroptosis.
1.1. Iron Metabolism and Lipid Peroxidation
Ferroptosis requires iron overload. The iron in the circulation of the human body exists in the form of Fe3+. Fe3+ is imported into the cell by transferrin and transferrin receptor 1 (TfR1) [6][7] then reduced to Fe2+ in the lysosome and, finally, released to the labile iron pool through the divalent metal 9 transporter 1 (DMT1) or zinc iron regulatory protein family 8/14 (ZIP8/14) [8][9]. Excess iron in cells is usually stored in ferritin [10], and ferritin can be recognized by the specific cargo receptor NCOA4, which recruits ferritin to autophagosomes for lysosomal degradation and releases Fe2+ [10][11][12][13]. The released Fe2+ will generate ROS through the Fenton reaction, and then undergoes a peroxidation reaction with lipids to trigger ferroptosis [2]. Increased iron absorption and reduced iron storage can lead to iron overload. Therefore, inhibiting iron overload by iron chelating agents can inhibit ferroptosis, and silencing the iron metabolism master regulator, iron responsive element binding protein 2 (IREB2), can also reduce the sensitivity of cells to ferroptosis [1].
In addition to abnormal iron metabolism, lipid peroxidation is also an important factor leading to ferroptosis. Lipidomic analysis showed that phosphatidylethanolamines (PEs) containing arachidonic acid (AA) or adrenal acid (AdA) are the key membrane phospholipids, which can be oxidized to phospholipid hydroperoxides (PE-AA/AdA-OOH) through non-enzymatic reactions, thereby driving ferroptosis [14][15]. The main ROS accumulated in cells are superoxide radical anions (·O2−) and hydrogen peroxide (H2O2). In the presence of free iron, these ROS may be converted into hydroxyl radicals (HO˙), which are highly reactive to PUFAs that exist in a variety of cell membranes [16]. Free PUFAs, including AA, are oxidized through a catalytic pathway involving acyl-CoA synthetase long-chain family member 4 (ACSL4), lysophosphatidylcholine acyltransferase 3 (LPCAT3), and lipoxygenase (LOXs) [17][18]. ACSL4 and LPCAT3 are key regulators of PUFA-PL biosynthesis [15]. ACSL4 acetylates PUFA to form PUFA-CoA, and then LPCAT3 inserts PUFA-CoA into lysophospholipid to form PUFA-PL [19]. Enzymatic lipid peroxidation is mainly regulated by the LOX family. LOXs can also oxidize PUFAs into corresponding phospholipid hydroperoxides, among which LOX5 and LOX12/15 are involved in ferroptosis [20]. It has been reported that phosphatidylethanolamine binding protein 1 (PEBP1) can form a complex with LOX15 and act as a scaffold protein to positively regulate ferroptosis [21]. It is worth noting here that 15-hydroperoxy (Hp)-arachidonoyl-phosphatidylethanolamine (15-HpETE-PE) produced by the 15-LOX/PEBP1 complex can be eliminated by Ca2+-independent phospholipase A2β (iPLA2β) [22]. This indicates that iPLA2β can act as an anti-ferroptotic guardian to regulate the intracellular ferroptosis signaling pathway, which has an important regulatory role in neurodegenerative diseases [22].
In addition, nicotinamide adenine phosphate dinucleotide (NADPH)-dependent lipid peroxidation and the inactivation of GPX4-induced lipid peroxidation are also involved in ferroptosis [23].
1.2. The System Xc−/GSH/GPX4 Axis
The system Xc−/GSH/GPX4 axis is involved in counteracting the endogenous iron–lipid peroxidation-dependent cell death pathway. If any of them are affected, this can trigger the occurrence of cell ferroptosis [24]. The Xc− transporter system consists of a regulatory subunit, solute carrier family 3 member 2 (SLC3A2), and a catalytic subunit, solute carrier family 7 member 11 (SLC7A11), which promote the cellular uptake of cystine by exchange with glutamate [25]. The cystine absorbed into the cell is then reduced to cysteine by GSH or thioredoxin reductase 1 (TrxR1), which in turn can synthesize GSH [26]. GSH is the main antioxidant in mammalian cells and a cofactor of GPX4 [27]. GPX4 can remove lipid hydroperoxides in biological membranes and use GSH to reduce cytotoxic lipid peroxides to non-toxic lipid alcohols, thereby interfering with the lipid peroxidation chain reaction [28]. Therefore, blocking the intracellular cysteine level will directly affect the activity of GPX4 through GSH, thereby increasing the sensitivity of cells to ferroptosis.
2. Mechanism of Ferroptosis in Non-alcoholic Fatty Liver Disease (NAFLD)
2.1. Decreased GPX4 Activity
Ferroptosis can trigger the inflammatory response of simple fatty liver degeneration, and this promotes the occurrence and development of NASH. In 2019, Tsurusaki et al. found that in the NASH mouse model induced by choline deficiency and ethionine supplementation (CDE) feed, hepatocyte death was accompanied by an increase in the level of oxygenated phospholipid ethanolamine [29]. The researchers also observed that in the initial stage of the development of NAFLD in mice to NASH, hepatocyte ferroptosis precedes cell apoptosis, which in turn leads to liver damage, immune cell infiltration, and inflammation. The ferroptosis inhibitors (e.g., trolox or deferoxamine) can almost completely reverse the death of liver cells, inflammation, and lipid peroxidation in the initial disease model of NASH (Figure 1). It also suppresses the subsequent infiltration of immune cells and inflammatory reaction, thereby improving liver function [29]. This suggests that hepatic ferroptosis, as a trigger of inflammation and initiation of steatohepatitis, may be a therapeutic target to prevent the onset of steatohepatitis.
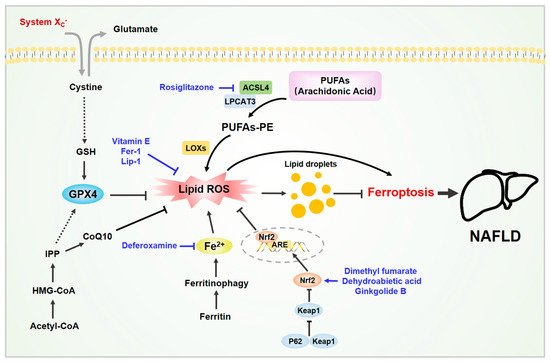
Figure 1. Ferroptosis in non-alcoholic fatty liver disease (NAFLD). Various molecules are involved in ferroptosis in NAFLD. The liver is the main organ for iron storage and lipid metabolism. The accumulation of iron and lipid peroxides in liver cells can induce cell ferroptosis, which in turn exacerbates NAFLD. Some active substances have been shown to inhibit hepatocyte ferroptosis and improve NAFLD by removing excess iron, neutralizing lipid peroxides, and activating the Nrf2 signaling pathway. Abbreviations: ACSL4, acylCoA synthetase long-chain family member 4; CoQ10, coenzyme Q10; GSH, glutathione; GPX4, glutathione peroxidase 4; IPP, isopentenyl pyrophosphate; LPCAT3, lysophosphatidylcholine acyltransferase 3. Black arrows indicate the activation modification, and black block lines indicate the inhibitory modification. Blue arrows indicate the activation process of activators, and blue block lines indicate the inhibition process of inhibitors. Black dashed arrows indicate the indirect process. Double arrows indicate the reverse transport process of glutamate and cystein.
Qi et al. also found that ferroptosis is a key factor in the development of NASH. Ferroptosis can aggravate the inflammatory response, oxidative stress, and cell damage in the early stages of NASH [30]. GPX4 can inhibit ferroptosis in patients with NASH. Specifically, RSL-3 (GPX4 inhibitor) promoted the progression of NASH by inducing ferroptosis in methionine and choline deficiency (MCD) diet-fed mice (serum biochemical levels and levels of hepatic steatosis and inflammation apoptosis were exacerbated), but sodium selenite (GPX4 activator) improved the severity of NASH [30]. Similarly, in the in vitro NASH model induced by palmitic acid, the regulation of ferroptosis mediated by GPX4 also affects the death of liver cells. Liver is the tissue with the highest expression of GPX4. GPX4 can protect the liver from lipid peroxidation, which is essential for liver function and liver cell survival [31].
In addition, it has been reported that some enzymes in cells can also affect ferroptosis by regulating GPX4. Thymosin β4 (Tβ4) is a multifunctional polypeptide that exists in a variety of nucleated cells. It can improve liver fibrosis and reduce inflammation [32]. Tβ4 can protect hepatocytes by inhibiting the GPX4-mediated ferroptosis pathway [33]. Enolase 3 (ENO3) is another enzyme that encodes the β-subunit of enolase that exists in the liver and in other organs [34]. ENO3 promoted the progression of NASH by negatively regulating ferroptosis via the elevation of GPX4 expression and lipid accumulation [35].
2.2. Iron Overload
2.3. Lipid Peroxidation
The latest research found that in the liver of mice that were fed with MCD feed, the metabolism of AA increased, iron accumulated, lipid ROS enhanced, and the levels of ironophilia-related genes (such as ACSL4, ALOX5AP, GPX4, and PTGS1) increased [37]. It is worth noting that Fe2+ is the key factor of ferroptosis, and AA is also the most common PUFA in ferroptosis. Therefore, the increase in Fe2+ levels and the increase in AA metabolism synergistically produce lipid peroxidation, which in turn promotes the development of NASH. Treatment of MCD diet mice with Fer-1 and Lip-1 significantly improved liver steatosis, liver damage, inflammation, and fibrosis in the mice, and it reduced the accumulation of lipid droplets and TG [37]. Finally, lipid ROS promotes liver steatosis by promoting the formation of lipid droplets. In terms of mechanism, inhibiting the expression of GPX4 increased the lipid droplets and lipid ROS in cells, while overexpression of GPX4 significantly inhibited the production of lipid droplets. This indicates that GPX4 plays an important role in the homeostasis of lipid droplets [37].
2.4. ACSL4 Induction
In addition to GPX4, ACSL4 also plays an important role in the ferroptosis of NAFLD and NASH. Wei et al. also observed ferroptosis in another risk factor of NASH, exposure to arsenic [38]. In both in vitro and in vivo models, exposure to arsenic could up-regulate the expression of ACSL4. However, both the ACSL4 inhibitor rosiglitazone (ROSI) and ACSL4 siRNA can suppress arsenic-induced ferroptosis. In addition, the use of Mitofusin 2 siRNA or IRE1α inhibitor reduced the content of 5-hydroxyeicosatetraenoic acid (5-HETE), which significantly alleviated NASH and ferroptosis [38].
2.5. Nrf2 Activation
The Nrf2-mediated antioxidant response plays a key role in the regulation of ferroptosis [39]. Nrf2 can promote the expression of downstream HO-1, GSH, and GPX4, thereby eliminating the accumulation of ROS in the liver and reducing the level of malondialdehyde (MDA) [40]. In addition, activation of the Nrf2 pathway in the obese mouse model can reduce liver lipid accumulation and significantly improve the NAFLD of the mice [41].
2.6. Others
A recent study found that enoyl coenzyme A hydratase 1 (ECH1), a key component in mitochondrial fatty acid β-oxidation, can reduce NASH in mice by inhibiting hepatic ferroptosis [42]. ECH1 expression was significantly increased in human NASH biopsy specimens and mice fed with MCD diets. ECH1 overexpression greatly alleviated liver steatosis, inflammation, fibrosis, and oxidative stress. Compared with untreated mice, ECH1-knockdown mice treated with Fer-1 showed a reduction in the NASH phenotype.
The Erk signaling pathway, of course, may be involved, and so this requires further in-depth research.
There is also an article studying the significance of microrna (miRNA) in the pathogenesis of fructose-induced NAFLD, and it found that fructose-induced oxidative damage can induce ferroptosis and that miR-33 can be used as a serological biomarker for fructose-induced NAFLD [43].
Overall, there are relatively few studies on ferroptosis in NAFLD at this stage. The role of ferroptosis in the various stages of NAFLD progression is worthy of further research and exploration, especially since there is no accurate choice for the treatment of NAFLD.
References
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072.
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191.
- Bochkov, V.N.; Oskolkova, O.V.; Birukov, K.G.; Levonen, A.L.; Binder, C.J.; Stöckl, J. Generation and biological activities of oxidized phospholipids. Antioxid. Redox Signal. 2010, 12, 1009–1059.
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17.
- Kapralov, A.A.; Yang, Q.; Dar, H.H.; Tyurina, Y.Y.; Anthonymuthu, T.S.; Kim, R.; St. Croix, C.M.; Mikulska-Ruminska, K.; Liu, B.; Shrivastava, I.H.; et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 2020, 16, 278–290.
- Yang, W.S.; Stockwell, B.R. Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem. Biol. 2008, 15, 234–245.
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308.
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem. Sci. 2016, 41, 274–286.
- Frazer, D.M.; Anderson, G.J. The regulation of iron transport. BioFactors 2014, 40, 206–214.
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428.
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032.
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 508, 105–109.
- Wang, Y.Q.; Chang, S.Y.; Wu, Q.; Gou, Y.J.; Jia, L.; Cui, Y.M.; Yu, P.; Shi, Z.H.; Wu, W.S.; Gao, G.; et al. The protective role of mitochondrial ferritin on erastin-induced ferroptosis. Front. Aging Neurosci. 2016, 8, 308.
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176.
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90.
- Berson, E.L.; Rosner, B.; Sandberg, M.A.; Weigel-DiFranco, C.; Moser, A.; Brockhurst, R.J.; Hayes, K.C.; Johnson, C.A.; Anderson, E.J.; Gaudio, A.R.; et al. Further evaluation of docosahexaenoic acid in patients with retinitis pigmentosa receiving vitamin A treatment: Subgroup analyses. Arch. Ophthalmol. 2004, 122, 1306–1314.
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98.
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975.
- Magtanong, L.; Ko, P.J.; Dixon, S.J. Emerging roles for lipids in non-apoptotic cell death. Cell Death Differ. 2016, 23, 1099–1109.
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425.
- Wenzel, S.E.; Tyurina, Y.Y.; Zhao, J.; St. Croix, C.M.; Dar, H.H.; Mao, G.; Tyurin, V.A.; Anthonymuthu, T.S.; Kapralov, A.A.; Amoscato, A.A.; et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 2017, 171, 628–641.
- Sun, W.Y.; Tyurin, V.A.; Mikulska-Ruminska, K.; Shrivastava, I.H.; Anthonymuthu, T.S.; Zhai, Y.J.; Pan, M.H.; Gong, H.B.; Lu, D.H.; Sun, J.; et al. Phospholipase iPLA2β averts ferroptosis by eliminating a redox lipid death signal. Nat. Chem. Biol. 2021, 17, 465–476.
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379.
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic. Biol. Med. 2019, 133, 144–152.
- Lo, M.; Wang, Y.Z.; Gout, P.W. The xc- cystine/glutamate antiporter: A potential target for therapy of cancer and other diseases. J. Cell. Physiol. 2008, 215, 593–602.
- Mandal, P.K.; Seiler, A.; Perisic, T.; Kölle, P.; Canak, A.B.; Förster, H.; Weiss, N.; Kremmer, E.; Lieberman, M.W.; Bannai, S.; et al. System xc- and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J. Biol. Chem. 2010, 285, 22244–22253.
- Kuganesan, N.; Dlamini, S.; McDaniel, J.; Tillekeratne, V.L.M.; Taylor, W.R. Identification and initial characterization of a potent inhibitor of ferroptosis. J. Cell. Biochem. 2021, 122, 413–424.
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-Lipoxygenase Dependent- and AIF-Mediated Cell Death. Cell Metab. 2008, 8, 237–248.
- Tsurusaki, S.; Tsuchiya, Y.; Koumura, T.; Nakasone, M.; Sakamoto, T.; Matsuoka, M.; Imai, H.; Yuet-Yin Kok, C.; Okochi, H.; Nakano, H.; et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 2019, 10, 449.
- Qi, J.; Kim, J.W.; Zhou, Z.; Lim, C.W.; Kim, B. Ferroptosis Affects the Progression of Nonalcoholic Steatohepatitis via the Modulation of Lipid Peroxidation–Mediated Cell Death in Mice. Am. J. Pathol. 2020, 190, 68–81.
- Carlson, B.A.; Tobe, R.; Yefremova, E.; Tsuji, P.A.; Hoffmann, V.J.; Schweizer, U.; Gladyshev, V.N.; Hatfield, D.L.; Conrad, M. Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Biol. 2016, 9, 22–31.
- Kim, J.; Jung, Y. Thymosin Beta 4 Is a Potential Regulator of Hepatic Stellate Cells. In Vitamins and Hormones; Elsevier: Amsterdam, The Netherlands, 2016; Volume 102, pp. 121–149. ISBN 9780128048184.
- Zhu, Z.; Zhang, Y.; Huang, X.; Can, L.; Zhao, X.; Wang, Y.; Xue, J.; Cheng, M.; Zhu, L. Thymosin beta 4 alleviates non-alcoholic fatty liver by inhibiting ferroptosis via up-regulation of GPX4. Eur. J. Pharmacol. 2021, 908, 174351.
- Wu, J.; Zhou, D.; Deng, C.; Wu, X.; Long, L.; Xiong, Y. Characterization of porcine ENO3: Genomic and cDNA structure, polymorphism and expression. Genet. Sel. Evol. 2008, 40, 563–579.
- Lu, D.; Xia, Q.; Yang, Z.; Gao, S.; Sun, S.; Luo, X.; Li, Z.; Zhang, X.; Han, S.; Li, X.; et al. ENO3 promoted the progression of NASH by negatively regulating ferroptosis via elevation of GPX4 expression and lipid accumulation. Ann. Transl. Med. 2021, 9, 661.
- Valenti, L.; Moscatiello, S.; Vanni, E.; Fracanzani, A.L.; Bugianesi, E.; Fargion, S.; Marchesini, G. Venesection for non-alcoholic fatty liver disease unresponsive to lifestyle counselling-a propensity score-adjusted observational study. QJM 2011, 104, 141–149.
- Li, X.; Wang, T.X.; Huang, X.; Li, Y.; Sun, T.; Zang, S.; Guan, K.L.; Xiong, Y.; Liu, J.; Yuan, H.X. Targeting ferroptosis alleviates methionine-choline deficient (MCD)-diet induced NASH by suppressing liver lipotoxicity. Liver Int. 2020, 40, 1378–1394.
- Wei, S.; Qiu, T.; Wang, N.; Yao, X.; Jiang, L.; Jia, X.; Tao, Y.; Zhang, J.; Zhu, Y.; Yang, G.; et al. Ferroptosis mediated by the interaction between Mfn2 and IREα promotes arsenic-induced nonalcoholic steatohepatitis. Environ. Res. 2020, 188, 109824.
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107.
- Gao, G.; Xie, Z.; Li, E.-W.; Yuan, Y.; Fu, Y.; Wang, P.; Zhang, X.; Qiao, Y.; Xu, J.; Hölscher, C.; et al. Dehydroabietic acid improves nonalcoholic fatty liver disease through activating the Keap1/Nrf2-ARE signaling pathway to reduce ferroptosis. J. Nat. Med. 2021, 75, 540–552.
- Yang, Y.; Chen, J.; Gao, Q.; Shan, X.; Wang, J.; Lv, Z. Study on the attenuated effect of Ginkgolide B on ferroptosis in high fat diet induced nonalcoholic fatty liver disease. Toxicology 2020, 445, 152599.
- Liu, B.; Yi, W.; Mao, X.; Yang, L.; Rao, C. Enoyl coenzyme A hydratase 1 alleviates nonalcoholic steatohepatitis in mice by suppressing hepatic ferroptosis. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E925–E937.
- Pan, J.H.; Cha, H.; Tang, J.; Lee, S.; Lee, S.H.; Le, B.; Redding, M.C.; Kim, S.; Batish, M.; Kong, B.C.; et al. The role of microRNA-33 as a key regulator in hepatic lipogenesis signaling and a potential serological biomarker for NAFLD with excessive dietary fructose consumption in C57BL/6N mice. Food Funct. 2021, 12, 656–667.
More
Information
Subjects:
Hematology; Cell Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
990
Revisions:
2 times
(View History)
Update Date:
20 Dec 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No