 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hideyuki Nakanishi | + 3109 word(s) | 3109 | 2021-11-08 09:31:04 | | | |
| 2 | Jason Zhu | Meta information modification | 3109 | 2021-12-17 02:15:31 | | |
Video Upload Options
Synthetic mRNAs, which are produced by in vitro transcription, have been recently attracting attention because they can express any transgenes without the risk of insertional mutagenesis. Protein-based translational regulation systems enable the context-dependent production of therapeutic proteins and have the potential to further improve the efficacy and safety of synthetic mRNAs.
1. Introduction
During gene expression, genes are first transcribed from DNA to messenger RNA (mRNA) and then translated from mRNA to protein. Thus, researchers can make cells express exogenous genes by transfecting either DNA or mRNA. Although DNA transfection is the standard method for transgene expression, it can cause insertional mutagenesis of endogenous genes, which is a major drawback in medical applications. In contrast, synthetic mRNAs, which are produced by in vitro transcription, can be used for transgene expression without the risk of insertional mutagenesis. Thus, synthetic mRNAs have recently been attracting attention as tools for gene therapy, cellular reprogramming, and vaccine development [1].
Nevertheless, context-dependent regulation of transgene expression is more difficult in synthetic mRNA transfection than in DNA transfection. Upon DNA transfection, transgene expression can be tuned using transcriptional regulatory sequences, such as drug-inducible or tissue-specific promoters. On the other hand, such transcriptional regulation cannot apply to synthetic mRNA transfection as they must be transcribed in vitro prior to transfection. As some therapeutic genes may cause adverse effects when they are expressed in inappropriate contexts, context-dependent transgene regulation is helpful to cope with low adverse effects and high therapeutic efficacy. For example, cell-selective expression of pro-apoptotic genes [2][3][4][5][6] may enable cancer cell elimination without damaging healthy cells. Similarly, cardiomyocyte-selective expression of proliferation-promoting genes can be helpful to cope with myocardial regeneration without elevating fibrosis and immune response [7]. Thus, the development of translational regulation systems will make synthetic mRNAs more valuable.
One promising approach is to develop protein-based systems for translational regulation of synthetic mRNAs. There are multiple advantages to protein-based translational regulation systems. First, when proteins are used as translational regulators, the regulators themselves can be translated from synthetic mRNAs, such that all components necessary for regulation can be delivered as synthetic mRNAs. Second, compared to ribozyme-embedded mRNAs [8] or caged mRNAs [9], mRNAs composing protein-based regulation systems have less concern regarding the alteration of mRNA properties during synthesis or storage, such as self-cleavage or decaging. Finally, in protein-based translational regulation systems, the first output protein can be used as the input for the second regulation, allowing layered gene circuits for sophisticated regulation ( Figure 1 ).
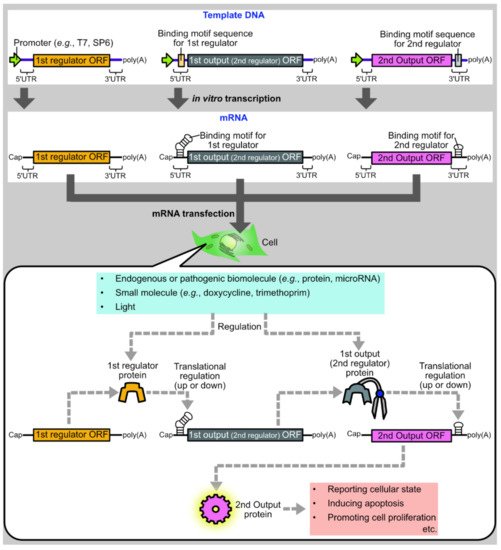
2. Basic Protein Modules for Protein-Based Translational Regulation Systems
2.1. Binding to Target mRNAs
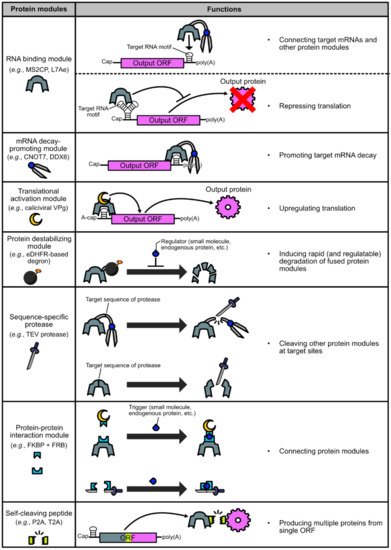
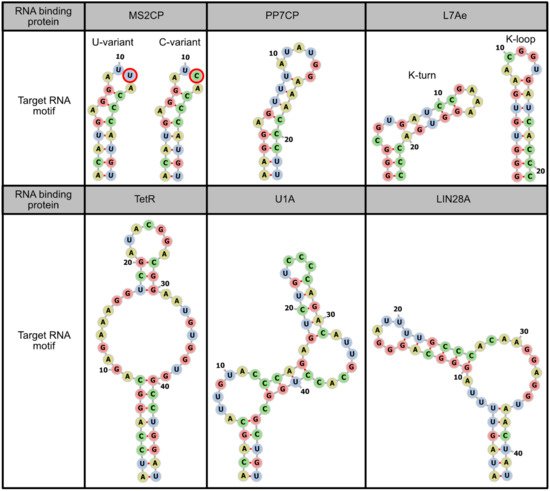
2.2. Promoting Target mRNA Decay
2.3. Activating Target mRNA Translation
2.4. Destabilizing Proteins
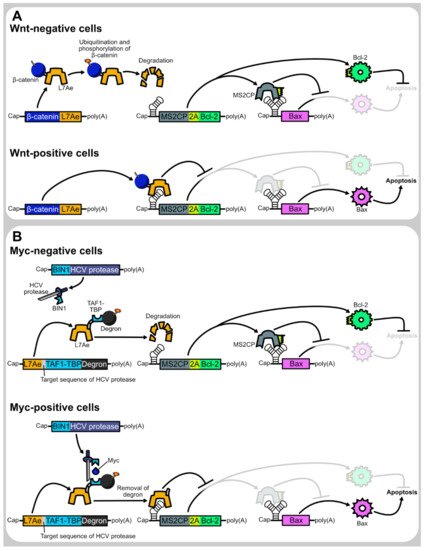
2.5. Cleaving Proteins
2.6. Combining Separate Proteins
2.7. Producing Multiple Proteins from a Single ORF
References
- Gómez-Aguado, I.; Rodríguez-Castejón, J.; Vicente-Pascual, M.; Rodríguez-Gascón, A.; Solinís, M.Á.; del Pozo-Rodríguez, A. Nanomedicines to Deliver MRNA: State of the Art and Future Perspectives. Nanomaterials 2020, 10, 364.
- Wroblewska, L.; Kitada, T.; Endo, K.; Siciliano, V.; Stillo, B.; Saito, H.; Weiss, R. Mammalian Synthetic Circuits with RNA Binding Proteins for RNA-Only Delivery. Nat. Biotechnol. 2015, 33, 839–841.
- Matsuura, S.; Ono, H.; Kawasaki, S.; Kuang, Y.; Fujita, Y.; Saito, H. Synthetic RNA-Based Logic Computation in Mammalian Cells. Nat. Commun. 2018, 9, 4847.
- Yang, J.; Ding, S. Engineering L7Ae for RNA-Only Delivery Kill Switch Targeting CMS2 Type Colorectal Cancer Cells. ACS Synth. Biol. 2021, 10, 1095–1105.
- Nakanishi, H.; Saito, H. Caliciviral Protein-Based Artificial Translational Activator for Mammalian Gene Circuits with RNA-Only Delivery. Nat. Commun. 2020, 11, 1297.
- Yang, J.; Ding, S. Chimeric RNA-Binding Protein-Based Killing Switch Targeting Hepatocellular Carcinoma Cells. Mol. Ther. - Nucleic Acids 2021, 25, 683–695.
- Magadum, A.; Singh, N.; Kurian, A.A.; Munir, I.; Mehmood, T.; Brown, K.; Sharkar, M.T.K.; Chepurko, E.; Sassi, Y.; Oh, J.G.; et al. Pkm2 Regulates Cardiomyocyte Cell Cycle and Promotes Cardiac Regeneration. Circulation 2020, 141, 1249–1265.
- Yokobayashi, Y. Aptamer-Based and Aptazyme-Based Riboswitches in Mammalian Cells. Curr. Opin. Chem. Biol. 2019, 52, 72–78.
- Zhang, D.; Zhou, C.Y.; Busby, K.N.; Alexander, S.C.; Devaraj, N.K. Light-Activated Control of Translation by Enzymatic Covalent MRNA Labeling. Angew. Chem. 2018, 130, 2872–2876.
- Stripecke, R.; Oliveira, C.C.; McCarthy, J.E.; Hentze, M.W. Proteins Binding to 5’ Untranslated Region Sites: A General Mechanism for Translational Regulation of MRNAs in Human and Yeast Cells. Mol. Cell. Biol. 1994, 14, 5898–5909.
- Endo, K.; Stapleton, J.A.; Hayashi, K.; Saito, H.; Inoue, T. Quantitative and Simultaneous Translational Control of Distinct Mammalian MRNAs. Nucleic Acids Res. 2013, 41, e135.
- Cella, F.; Wroblewska, L.; Weiss, R.; Siciliano, V. Engineering Protein-Protein Devices for Multilayered Regulation of MRNA Translation Using Orthogonal Proteases in Mammalian Cells. Nat. Commun. 2018, 9, 4392.
- Parr, C.J.C.; Wada, S.; Kotake, K.; Kameda, S.; Matsuura, S.; Sakashita, S.; Park, S.; Sugiyama, H.; Kuang, Y.; Saito, H. N1-Methylpseudouridine Substitution Enhances the Performance of Synthetic MRNA Switches in Cells. Nucleic Acids Res. 2020, 48, e35.
- Liu, K.; Yang, J.; Ding, S.; Gao, Y. Daisy Chain Topology Based Mammalian Synthetic Circuits for RNA-Only Delivery. ACS Synth. Biol. 2020, 9, 269–282.
- Paek, K.Y.; Hong, K.Y.; Ryu, I.; Park, S.M.; Keum, S.J.; Kwon, O.S.; Jang, S.K. Translation Initiation Mediated by RNA Looping. Proc. Natl. Acad. Sci. 2015, 112, 1041–1046.
- Hosoda, N.; Kim, Y.K.; Lejeune, F.; Maquat, L.E. CBP80 Promotes Interaction of Upf1 with Upf2 during Nonsense-Mediated MRNA Decay in Mammalian Cells. Nat. Struct. Mol. Biol. 2005, 12, 893–901.
- Kim, Y.K.; Furic, L.; DesGroseillers, L.; Maquat, L.E. Mammalian Staufen1 Recruits Upf1 to Specific MRNA 3′UTRs so as to Elicit MRNA Decay. Cell 2005, 120, 195–208.
- Lykke-Andersen, J.; Shu, M.-D.; Steitz, J.A. Communication of the Position of Exon-Exon Junctions to the MRNA Surveillance Machinery by the Protein RNPS1. Science 2001, 293, 1836–1839.
- Lykke-Andersen, J.; Shu, M.-D.; Steitz, J.A. Human Upf Proteins Target an MRNA for Nonsense-Mediated Decay When Bound Downstream of a Termination Codon. Cell 2000, 103, 1121–1131.
- Nakanishi, H.; Yoshii, T.; Kawasaki, S.; Hayashi, K.; Tsutsui, K.; Oki, C.; Tsukiji, S.; Saito, H. Light-Controllable RNA-Protein Devices for Translational Regulation of Synthetic MRNAs in Mammalian Cells. Cell Chem. Biol. 2021, 28, 662–674.e5.
- Ono, H.; Kawasaki, S.; Saito, H. Orthogonal Protein-Responsive MRNA Switches for Mammalian Synthetic Biology. ACS Synth. Biol. 2020, 9, 169–174.
- Stapleton, J.A.; Endo, K.; Fujita, Y.; Hayashi, K.; Takinoue, M.; Saito, H.; Inoue, T. Feedback Control of Protein Expression in Mammalian Cells by Tunable Synthetic Translational Inhibition. ACS Synth. Biol. 2012, 1, 83–88.
- Wagner, T.E.; Becraft, J.R.; Bodner, K.; Teague, B.; Zhang, X.; Woo, A.; Porter, E.; Alburquerque, B.; Dobosh, B.; Andries, O.; et al. Small-Molecule-Based Regulation of RNA-Delivered Circuits in Mammalian Cells. Nat. Chem. Biol. 2018, 14, 1043–1050.
- Shen, C.-C.; Hsu, M.-N.; Chang, C.-W.; Lin, M.-W.; Hwu, J.-R.; Tu, Y.; Hu, Y.-C. Synthetic Switch to Minimize CRISPR Off-Target Effects by Self-Restricting Cas9 Transcription and Translation. Nucleic Acids Res. 2019, 47, e13.
- Mc Cafferty, S.; De Temmerman, J.; Kitada, T.; Becraft, J.R.; Weiss, R.; Irvine, D.J.; Devreese, M.; De Baere, S.; Combes, F.; Sanders, N.N. In Vivo Validation of a Reversible Small Molecule-Based Switch for Synthetic Self-Amplifying MRNA Regulation. Mol. Ther. J. Am. Soc. Gene Ther. 2021, 29, 1164–1173.
- Goldfless, S.J.; Belmont, B.J.; de Paz, A.M.; Liu, J.F.; Niles, J.C. Direct and Specific Chemical Control of Eukaryotic Translation with a Synthetic RNA–Protein Interaction. Nucleic Acids Res. 2012, 40, e64.
- Kawasaki, S.; Fujita, Y.; Nagaike, T.; Tomita, K.; Saito, H. Synthetic MRNA Devices That Detect Endogenous Proteins and Distinguish Mammalian Cells. Nucleic Acids Res. 2017, 45, e117.
- Lowary, P.T.; Uhlenbeck, O.C. An RNA Mutation That Increases the Affinity of an RNA-Protein Interaction. Nucleic Acids Res. 1987, 15, 10483–10493.
- Nagai, K.; Oubridge, C.; Ito, N.; Avis, J.; Evans, P. The RNP Domain: A Sequence-Specific RNA-Binding Domain Involved in Processing and Transport of RNA. Trends Biochem. Sci. 1995, 20, 235–240.
- Kerpedjiev, P.; Hammer, S.; Hofacker, I.L. Forna (Force-Directed RNA): Simple and Effective Online RNA Secondary Structure Diagrams. Bioinformatics 2015, 31, 3377–3379.
- Mugridge, J.S.; Coller, J.; Gross, J.D. Structural and Molecular Mechanisms for the Control of Eukaryotic 5′–3′ MRNA Decay. Nat. Struct. Mol. Biol. 2018, 25, 1077–1085.
- Inada, T.; Makino, S. Novel Roles of the Multi-Functional CCR4-NOT Complex in Post-Transcriptional Regulation. Front. Genet. 2014, 5, 135.
- Pillai, R.S.; Artus, C.G.; Filipowicz, W. Tethering of Human Ago Proteins to MRNA Mimics the MiRNA-Mediated Repression of Protein Synthesis. RNA 2004, 10, 1518–1525.
- Babendure, J.R.; Babendure, J.L.; Ding, J.-H.; Tsien, R.Y. Control of Mammalian Translation by MRNA Structure near Caps. RNA 2006, 12, 851–861.
- Petrakova, O.; Volkova, E.; Gorchakov, R.; Paessler, S.; Kinney, R.M.; Frolov, I. Noncytopathic Replication of Venezuelan Equine Encephalitis Virus and Eastern Equine Encephalitis Virus Replicons in Mammalian Cells. J. Virol. 2005, 79, 7597–7608.
- Yoshioka, N.; Gros, E.; Li, H.-R.; Kumar, S.; Deacon, D.C.; Maron, C.; Muotri, A.R.; Chi, N.C.; Fu, X.-D.; Yu, B.D.; et al. Efficient Generation of Human IPSCs by a Synthetic Self-Replicative RNA. Cell Stem Cell 2013, 13, 246–254.
- Fukao, A.; Tomohiro, T.; Fujiwara, T. Translation Initiation Regulated by RNA-Binding Protein in Mammals: The Modulation of Translation Initiation Complex by Trans-Acting Factors. Cells 2021, 10, 1711.
- Herbert, T.P.; Brierley, I.; Brown, T.D. Identification of a Protein Linked to the Genomic and Subgenomic MRNAs of Feline Calicivirus and Its Role in Translation. J. Gen. Virol. 1997, 78, 1033–1040.
- Goodfellow, I.; Chaudhry, Y.; Gioldasi, I.; Gerondopoulos, A.; Natoni, A.; Labrie, L.; Laliberté, J.-F.; Roberts, L. Calicivirus Translation Initiation Requires an Interaction between VPg and EIF4E. EMBO Rep. 2005, 6, 968–972.
- Chaudhry, Y.; Nayak, A.; Bordeleau, M.-E.; Tanaka, J.; Pelletier, J.; Belsham, G.J.; Roberts, L.O.; Goodfellow, I.G. Caliciviruses Differ in Their Functional Requirements for EIF4F Components. J. Biol. Chem. 2006, 281, 25315–25325.
- Matsui, A.; Uchida, S.; Ishii, T.; Itaka, K.; Kataoka, K. Messenger RNA-Based Therapeutics for the Treatment of Apoptosis-Associated Diseases. Sci. Rep. 2015, 5, 15810.
- De Gregorio, E.; Baron, J.; Preiss, T.; Hentze, M.W. Tethered-Function Analysis Reveals That ElF4E Can Recruit Ribosomes Independent of Its Binding to the Cap Structure. RNA 2001, 7, 106–113.
- Banaszynski, L.A.; Chen, L.; Maynard-Smith, L.A.; Lisa Ooi, A.G.; Wandless, T.J. A Rapid, Reversible, and Tunable Method to Regulate Protein Function in Living Cells Using Synthetic Small Molecules. Cell 2006, 126, 995–1004.
- Nishimura, K.; Fukagawa, T.; Takisawa, H.; Kakimoto, T.; Kanemaki, M. An Auxin-Based Degron System for the Rapid Depletion of Proteins in Nonplant Cells. Nat. Methods 2009, 6, 917–922.
- Iwamoto, M.; Björklund, T.; Lundberg, C.; Kirik, D.; Wandless, T.J. A General Chemical Method to Regulate Protein Stability in the Mammalian Central Nervous System. Chem. Biol. 2010, 17, 981–988.
- Taxis, C.; Stier, G.; Spadaccini, R.; Knop, M. Efficient Protein Depletion by Genetically Controlled Deprotection of a Dormant N-Degron. Mol. Syst. Biol. 2009, 5, 267.
- Tözsér, J.; Tropea, J.E.; Cherry, S.; Bagossi, P.; Copeland, T.D.; Wlodawer, A.; Waugh, D.S. Comparison of the Substrate Specificity of Two Potyvirus Proteases. FEBS J. 2005, 272, 514–523.
- Pethe, M.A.; Rubenstein, A.B.; Khare, S.D. Data-Driven Supervised Learning of a Viral Protease Specificity Landscape from Deep Sequencing and Molecular Simulations. Proc. Natl. Acad. Sci. USA 2019, 116, 168–176.
- Bayle, J.H.; Grimley, J.S.; Stankunas, K.; Gestwicki, J.E.; Wandless, T.J.; Crabtree, G.R. Rapamycin Analogs with Differential Binding Specificity Permit Orthogonal Control of Protein Activity. Chem. Biol. 2006, 13, 99–107.
- Liberles, S.D.; Diver, S.T.; Austin, D.J.; Schreiber, S.L. Inducible Gene Expression and Protein Translocation Using Nontoxic Ligands Identified by a Mammalian Three-Hybrid Screen. Proc. Natl. Acad. Sci. USA 1997, 94, 7825–7830.
- Kennedy, M.J.; Hughes, R.M.; Peteya, L.A.; Schwartz, J.W.; Ehlers, M.D.; Tucker, C.L. Rapid Blue-Light–Mediated Induction of Protein Interactions in Living Cells. Nat. Methods 2010, 7, 973–975.
- Dagliyan, O.; Krokhotin, A.; Ozkan-Dagliyan, I.; Deiters, A.; Der, C.J.; Hahn, K.M.; Dokholyan, N.V. Computational Design of Chemogenetic and Optogenetic Split Proteins. Nat. Commun. 2018, 9, 4042.
- Kim, J.H.; Lee, S.-R.; Li, L.-H.; Park, H.-J.; Park, J.-H.; Lee, K.Y.; Kim, M.-K.; Shin, B.A.; Choi, S.-Y. High Cleavage Efficiency of a 2A Peptide Derived from Porcine Teschovirus-1 in Human Cell Lines, Zebrafish and Mice. PLoS ONE 2011, 6, e18556.

