Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Yu, L. Adipose Lipolysis. Encyclopedia. Available online: https://encyclopedia.pub/entry/17112 (accessed on 24 June 2026).
Yu L. Adipose Lipolysis. Encyclopedia. Available at: https://encyclopedia.pub/entry/17112. Accessed June 24, 2026.
Yu, Liqing. "Adipose Lipolysis" Encyclopedia, https://encyclopedia.pub/entry/17112 (accessed June 24, 2026).
Yu, L. (2021, December 14). Adipose Lipolysis. In Encyclopedia. https://encyclopedia.pub/entry/17112
Yu, Liqing. "Adipose Lipolysis." Encyclopedia. Web. 14 December, 2021.
Copy Citation
The heart primarily uses fatty acids as energy substrates. Adipose lipolysis is a major source of fatty acids, particularly under stress conditions. Emerging evidence suggests a bidirectional communication between the heart and adipose tissue. The entry is the first to define how adipose CGI58-mediated lipolysis influences cardiac remodeling and function. The adipose-heart axis may be targeted for the management of cardiac dysfunction.
Abhd5
adipose lipolysis
cardiac energy substrates and remodeling
cardiac hypertrophy and dysfunction
1. Introduction
Obesity and associated metabolic complications, including insulin resistance, type 2 diabetes, dyslipidemia, fatty liver disease, and cardiometabolic dysfunction, are a global health burden. Obesity is characterized by increased visceral fat, a type of adipose tissue that has the most detrimental metabolic effects. Adipose tissue can be classified into two major subtypes: white adipose tissue (WAT) and brown adipose tissue (BAT). WAT stores excess energy as triglycerides (TGs) in the unilocular cytosolic lipid droplets of white adipocytes. BAT dissipates metabolic energy as heat through the mitochondrial uncoupling protein 1 (Ucp-1) for nonshivering thermogenesis [1], though BAT also stores TGs in the multilocular small cytosolic lipid droplets of brown adipocytes. Another subtype of adipose tissue is called beige adipose tissue, which resides in the classical WAT depots but has features between WAT and BAT. Beige adipose tissue can be recruited to WAT depots by cold exposure and some agents, and this process is called white adipose browning [2][3]. Human adults possess substantial beige fat, though lacking grossly identifiable brown fat pads as seen in human infants and rodents [4][5][6]. Since BAT and beige fat convert metabolic energy into heat, agents and interventions recruiting BAT and beige fat may help combat obesity and associated metabolic disorders.
The hydrolytic process that mobilizes cytosolic lipid droplet TG stores is called intracellular lipolysis [7]. During starvation or increased energy demands, intracellular lipolysis is stimulated, resulting in sequential cleavage of three fatty acyl chains from a TG molecule by three hydrolytic enzymes, adipose triglyceride lipase (ATGL), hormone-sensitive lipase, and monoglyceride lipase. ATGL is the rate-limiting enzyme and requires the coactivator CGI-58 to fully function [8][9]. The metabolic fate of fatty acids (FAs) released from lipid droplet lipolysis differs between WAT and BAT. FAs and glycerol from WAT lipolysis are released into the blood circulation to serve, respectively, as energy fuels for vital organs such as the heart and as the biosynthetic backbone for metabolic pathways such as hepatic gluconeogenesis. On the other hand, the lipolytic products in BAT are predominantly utilized by mitochondria within the same cells for heat generation. Despite having seemingly opposite functions, WAT and BAT coordinate in many aspects. For example, when BAT lipolysis is disrupted, animals rely on WAT lipolysis to maintain body temperature during fasting and cold stress [10][11][12].
Coordination exists not only among different adipose subtypes but also between adipose and non-adipose tissues. Recent research on adipose biology has discovered that adipose tissue can function as an endocrine organ via the secretion of adipokines, extracellular vesicles, special lipids, non-coding RNAs, and metabolites [13][14][15]. These versatile functions of adipose tissue place it at the crossroad of organ-organ communications in energy metabolism, normal physiology, and disease pathophysiology. In adults, normal cardiac function relies predominantly on the continuous supply of FAs as energy substrates [16]. Exogenous FAs delivered to cardiomyocytes in the heart are mainly derived from adipose lipolysis and/or hydrolysis of TGs in TG-rich lipoproteins by lipoprotein lipase (i.e., intravascular lipolysis) [7][17]. It has been shown that adipose-specific deletion of ATGL alters cardiac lipidome and prevents cardiac hypertrophy and heart failure in chow-fed mice under the transverse aortic constriction-induced pressure overload [18]. When these animals are subjected to chronic exercise, they show increased use of glucose in the heart as measured by 18F-fluorodeoxyglucose [19]. Adipose deletion of ATGL or its coactivator CGI-58 renders mice cold sensitive in the fasted state [10][12]. It remains completely unknown how the hearts of these animals respond to thermal stress such as cold exposure, a condition often used in thermoregulation and energy metabolism research and experienced by people in the winter and living in the northern hemisphere. Army soldiers are also often exposed to prolonged cold stress during training and on the battlefield. Cold stress has been linked to increasing cardiovascular events [20], but the underlying mechanisms remain elusive. The heart is an integral part of the thermoregulatory system since it disseminates heated blood to maintain body temperature. Additionally, the heart secretes atrial and brain-type natriuretic peptides (ANP and BNP) that have been shown to promote lipolysis and thermogenesis in the adipose tissue [21][22]. Cold stress is known to stimulate the sympathetic nervous system whose activation is the most powerful inducer of adipose lipolysis [1].
2. Selective Inactivation of CGI-58 in Adipose Tissue Reduces Blood Free Fatty Acids and Increases Whole-Body Glucose Utilization
We have previously shown that FAT-KO relative to control mice gain similar weight on a regular chow or high-fat diet (HFD) and are completely resistant to the β-adrenergic receptor agonist isoproterenol-stimulated lipolysis as evidenced by the failed release of glycerol and FFAs into the blood circulation [10]. The WAT explant from FAT-KO mice was resistant to the isoproterenol-stimulated release of glycerol into the culture medium (Figure 1A). Importantly, chronic deficiency in adipose lipolysis lowered serum FFA levels under both fed and fasted states (Figure 1B,C). Considering the Randle (glucose-FA) cycle of metabolic fuel competition [23], we hypothesized that mice lacking adipose lipolysis are metabolically reprogrammed to utilize more glucose, especially under increased energy demanding conditions such as cold stress. To test this hypothesis, we placed the animals in a cold chamber. As expected, cold exposure increased total energy expenditure (TEE) in both genotypes (Figure 1D). The respiratory exchange ratio (RER) is an indicator of metabolic fuel sources. A higher value is indicative of carbohydrates versus fat as the preferred energy substrates. Consistently with our hypothesis, the RER was significantly higher in FAT-KO mice than the controls during cold exposure (Figure 1D). Increased use of carbohydrates is expected to improve glucose disposal. Indeed, FAT-KO mice compared to the controls showed better tolerance to glucose and were more sensitive to insulin-stimulated glucose disposal at normal housing temperature (Figure 1E). Despite no changes in serum glucose levels, serum insulin levels were consistently lower in FAT-KO mice than the control mice, resulting in a significant reduction in the homeostatic model assessment of insulin resistance index in FAT-KO mice (Figure 1F). Interestingly, a bolus of glucose injection cannot raise blood glucose levels in the FAT-KO mice during acute cold exposure (Figure 1G), suggesting an instant and complete tissue disposal of exogenous glucose by FAT-KO mice.
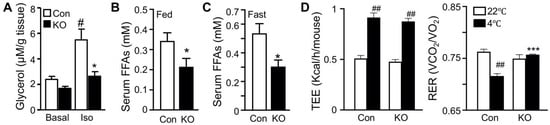
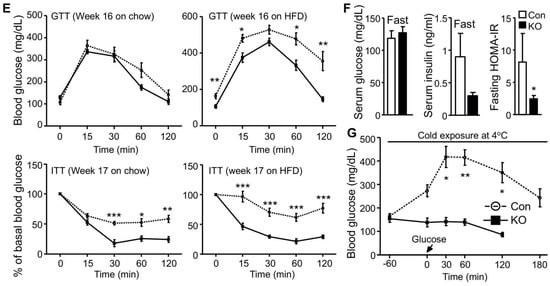
Figure 1. Deletion of adipose CGI-58 reduces serum FFA concentrations and increases whole-body glucose utilization. (A) Ex vivo lipolysis of iWAT explants from 14-week-old chow-fed mice (n = 5/group). Glycerol concentrations in the medium of cultured iWAT explants were measured after a 2 h incubation at 37 °C in the absence (Basal) or presence of isoproterenol (Iso). (B,C) Serum concentrations of FFAs in the 15-week-old HFD-fed mice in the fed state (B) and in the 17-week-old HFD-fed mice after overnight fasting (C) (n = 5–6/group). (D) The respiratory exchange ratio (RER) and the total energy expenditure (TEE) in 23-week-old FAT-KO and control mice fed the HFD ad libitum at room temperature and during cold exposure. The RER and TEE shown represent the data continuously collected for 3 h immediately before and 1 h after the switch of the housing temperature from 22 °C to 4 °C (n = 4–6/group). (E) Insulin and glucose tolerance tests in mice fed a chow diet (n = 5–6/group) or HFD starting at six weeks of age (n = 7–8/group) at room temperature. (F) Serum levels of insulin, glucose, and fasting homeostatic model assessment of insulin resistance (HOMA-IR) indexes in 17-week-old HFD-fed mice (n = 5–6/group). (G) Blood glucose levels of 11-week-old HFD-fed mice during acute cold exposure (n = 4–5/group). The mice were fasted for ~6 h during the daytime cycle and then exposed to 4 °C for 1 h prior to intraperitoneal injection of a bolus of glucose at 1.5 g/kg BW while remaining at 4 °C for the time indicated. * p < 0.05, ** p < 0.01, and *** p < 0.001 vs. genotype; # p < 0.05 vs. treatment; ## p < 0.001 vs. housing temperature by two-tailed unpaired Student’s t-test.
3. CGI-58 Deletion in the Adipose Tissue Increases Glucose Uptake in the Heart
Increased glucose disposal suggests enhanced tissue glucose uptake. Adipose and muscle tissues are major sites responsible for glucose disposal in peripheral organs. We, therefore, measured glucose uptake in these tissues by using the radioactive 2-deoxyglucose as a tracer. Under the insulin-stimulated condition, the hearts of FAT-KO mice relative to the control mice showed a >2-fold increase in glucose uptake (Figure 2A). Remarkably, cardiac glucose uptake increased >10-fold during acute cold exposure without insulin administration (Figure 2B). Relative to the heart, other tissues including interscapular brown adipose tissue, inguinal WAT, epididymal WAT, gastrocnemius muscle, and quadriceps muscle, showed lower glucose uptake per tissue weight under both insulin-stimulated and cold conditions, regardless of genotypes. Under the insulin-stimulated condition, gastrocnemius muscle and epididymal WAT had increased and decreased glucose uptake, respectively, in FAT-KO mice relative to control mice. Among the tissues examined, the heart was the only one showing increased glucose uptake per tissue wet weight in FAT-KO mice compared to the controls during cold stimulation.
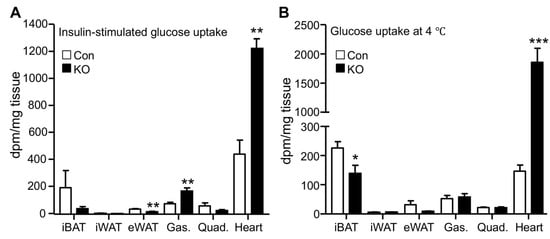
Figure 2. Adipose CGI-58 deficiency increases cardiac glucose uptake. (A) Tissue glucose uptake stimulated by insulin in 28-week-old HFD-fed mice housed at room temperature (n = 4–6/group). (B) Tissue glucose uptake in 20-week-old HFD-fed mice during acute cold exposure (n = 4–5/group). * p < 0.05, ** p < 0.01, and *** p < 0.001 vs. genotype by two-tailed unpaired Student’s t-test. iBAT, interscapular brown adipose tissue; iWAT, inguinal subcutaneous white adipose tissue; eWAT, epididymal white adipose tissue; Gas., gastrocnemius muscle; and Quad., quadriceps muscle.
4. Adipose CGI-58 Deficiency Increases Cardiac Expression of Natriuretic Peptides
Normal adult hearts preferentially use FFAs as metabolic fuels. The increase in cardiac glucose uptake observed in FAT-KO mice prompted us to examine whether there was remodeling in the heart. Since cardiac remodeling is a chronic process, we measured atrial and brain natriuretic peptides (ANP and BNP), the two sensitive markers of cardiac remodeling and functional changes [24], in mice that were housed in normal room temperature. Indeed, there was a significant increase in cardiac mRNAs for both ANP and BNP (Figure 3A). Since cold stress-induced a >10-fold increase in cardiac glucose uptake (Figure 2B), we were interested in cold-associated cardiac remodeling. We subjected the mice to a long-term cold exposure regimen (7 days cold acclimation followed by 32 days of cold exposure at 6 °C) and then measured cardiac levels of mRNAs for ANP and BNP. The ad libitum fed FAT-KO mice at the age indicated can survive long-term cold exposure and maintain body temperature (Figure 3B). Interestingly, the body temperature was maintained at a higher level in the ad libitum-fed FAT-KO than control mice during chronic cold exposure. About one month after cold exposure, FAT-KO mice showed a substantial increase in cardiac levels of mRNAs for ANP and BNP (Figure 3C), and their plasma concentrations of ANP were also increased (Figure 3D). To determine whether these increases in ANP and BNP were associated with alterations in cardiac functions, we performed echocardiography. Interestingly, FAT-KO mice displayed a significant reduction in fractional shortening of LV posterior wall (FS-PW), though LV ejection fraction (LVEF) was not reduced (Figure 3E).
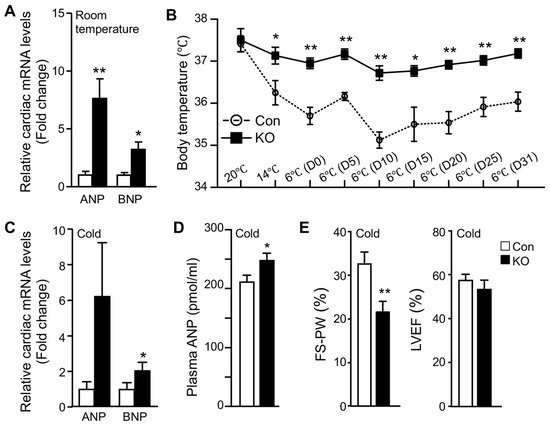
Figure 3. Adipose CGI-58 deficiency increases cardiac mRNA expression of natriuretic peptides and plasma ANP concentrations. (A) Relative mRNA levels of ANP and BNP in the hearts of 14-week-old HFD-fed mice housed at room temperature (n = 4–5/group). (B–E) Body temperature changes (B, n = 6–8), relative mRNA levels of natriuretic peptides (C, n = 5), plasma concentrations of ANP (D, n = 6–8), and echocardiography analysis (E, n = 6–8) of the HFD-fed mice that were subjected to cold acclimation (2 °C/day reduction in housing temperatures) for 7 days, followed by 32 days of cold exposure at 6 °C. * p < 0.05 and ** p < 0.01 vs. genotype by two-tailed unpaired Student’s t-test.
5. Adipose CGI-58 Deficiency Attenuates Cardiac Function during Cold Exposure
We speculated that high-fat feeding may have provided the heart with sufficient exogenous FFAs postprandially and slowed down the impact of adipose lipolysis deficiency on cardiac remodeling and function. In addition, a relatively long time is often needed for metabolic dysregulation to cause significant functional changes due to the generally large compensatory capacity of a heart. Given these, we measured cardiac functions in a cohort of middle-aged (10–11-months old) and chow-fed mice before and after 26 days of cold exposure (Figure 4). Interestingly, the heart rate was significantly higher in FAT-KO mice compared to the controls at the basal room temperature. Following the cold exposure, the heart rate was elevated in the control, but not KO mice. There was no significant difference in the heart rate between the two genotypes after cold exposure. The LV posterior wall thickness at end-diastole (LVPWd) increased in both genotypes following cold exposure, suggesting cold-induced LV hypertrophy. FAT-KO mice had an even greater response than control mice, causing a significant increase in LVPWd in these animals under cold stress. We did not observe any differences in the heart-to-body weight ratio following cold exposure (FAT-KO, 8.12% of BW vs. Control, 7.63% of BW), likely since the heart weight is generally not sensitive for LV hypertrophy in this model on a regular chow diet. There were no significant differences between the two genotypes in other echocardiographic parameters at the baseline (room temperature). Left ventricular fractional shortening (FS-LVD), LVEF, and stroke volume (SV) significantly increased in control mice following cold exposure, but these increases were not observed in FAT-KO mice. Following cold exposure, FS-LVD and LVEF were significantly lower in FAT-KO mice than controls. Therefore, it is evident that deletion of adipose CGI-58 disrupted the inotropic response of the heart to cold stress, which led to depressed LV systolic function. There were no significant changes in diastolic functions between the two genotypes or their responses to cold exposure (data not shown).
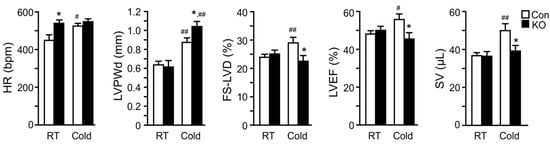
Figure 4. Deletion of adipose CGI-58 depresses cardiac responses to cold stress in chow-fed mice. Echocardiography analysis of cardiac structural and functional changes in 41–44-week-old chow-fed mice after cold acclimation (2 °C/day reduction in housing temperatures) for 7 days followed by housing at 6 °C for 26 days before analysis (n = 6–9/group). * p < 0.05 vs. genotype; # p < 0.05 and ## p < 0.01 vs. housing temperature by two-tailed unpaired Student’s t-test. HR, heart rate; LVPWd, left ventricular posterior wall thickness at end diastole; FS-LVD, left ventricular fractional shortening; LVEF, left ventricular ejection fraction; SV, stroke volume.
6. Adipose CGI-58 Deficiency Induces Pathological Remodeling of the Heart
To determine whether the depressed response of LV systolic functions to cold stress seen in FAT-KO mice was physiological or pathological, we first determined whether mitochondrial oxidative phosphorylation was affected in the FAT-KO mice since the heart as a major oxidative organ relies upon oxidative phosphorylation in mitochondria to generate energy and fulfill its vital function. We measured cardiac levels of representative proteins in five mitochondrial respiratory chain complexes at the early stage of cold exposure. Although no significant changes were observed for all of the representative proteins between the two genotypes at room temperature, levels of Complex I component NADH: ubiquinone oxidoreductase subunit B8 (NDUFB8), Complex II component succinate dehydrogenase complex iron sulfur subunit B (SDHB), and Complex III component ubiquinol-cytochrome C reductase core protein 2 (UQCRC2) were significantly reduced in FAT-KO mice under the cold stress (Figure 5A). Failed increases in cardiac mitochondrial respiratory chain proteins in the cold-exposed FAT-KO mice may not be able to meet the increased energy demand under chronic cold conditions, thereby causing cardiac remodeling. We, therefore, measured ventricular protein expression levels of genes related to pathological cardiac hypertrophy in FAT-KO mice after chronic cold exposure. There was a significant increase in collagen I and β-myosin heavy chain (MHC-β) and a significant decrease in sarcoplasmic reticulum Ca2+-ATPase (SERCA2) in FAT-KO mice compared to control mice (Figure 5B,C). This finding implies pathological remodeling of the heart in FAT-KO mice under cold stress.
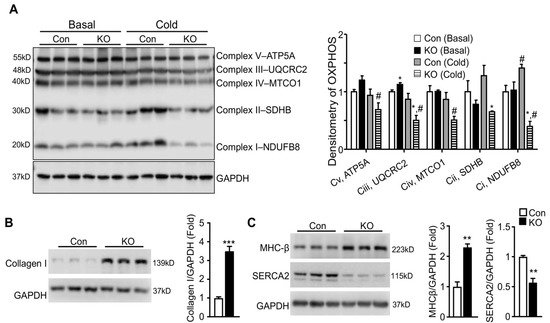
Figure 5. Deletion of adipose CGI-58 induces pathological remodeling of the heart. (A) Western blots and densitometry analysis of mitochondrial electron transport chain complex proteins in the hearts of 10–12-week-old chow-fed mice with or without cold exposure for seven days. (B,C) Western blots and densitometry analysis of biomarkers for pathological remodeling in the hearts of the mice described under Figure 4. * p < 0.05, ** p < 0.01, *** p < 0.001 vs. genotype; # p < 0.05 vs. housing temperature by two-tailed unpaired Student’s t-test. MTCO1, mitochondrially encoded cytochrome C oxidase 1; NDUFB8, NADH:ubiquinone oxidoreductase subunit B8; SDHB, succinate dehydrogenase complex iron sulfur subunit B (SDHB), UQCRC2, ubiquinol-cytochrome C reductase core protein 2.
7. Adipose CGI-58 Deficiency Increases Glucose Transporter Expression Levels and Cardiomyocytes in Hearts of Chow-Fed Mice after Chronic Cold Stress
In the heart, glucose uptake is mainly controlled by glucose transporter 1 (Glut1) and Glut4. Glut1 is responsible for constitutive uptake of glucose [25], whereas Glut4 mediates insulin-stimulated glucose uptake. Both glucose transporters can translocate to the cell surface to mediate glucose uptake. To determine whether adipose lipolysis deficiency altered the abundance and subcellular localization of cardiac Glut1 and Glut4 in mice after cold exposure, we first performed immunofluorescence studies by using antibodies against Glut1 and Glut4 as well as Wheat Germ Agglutinin (WGA) that stains cells’ surfaces. It was observed that the intensity of fluorescence for both Glut1 and Glut4 proteins was much stronger and there was more colocalization of Glut1 or Glut4 with WGA in the hearts of FAT-KO mice than the controls after cold stress (Figure 6A–C). Consistent with increased fluorescence intensity, the heart of FAT-KO mice also expressed more Glut1 and Glut4 proteins when measured by immunoblotting (Figure 6D). When observing the WGA-stained sections under the microscope, we noticed apparent enlargement of cardiomyocytes and therefore performed morphometric analysis. Indeed, there was a significant increase in the diameters of cardiomyocytes in FAT-KO mice (Figure 6E), suggesting the development of cardiac hypertrophy in these animals after chronic cold exposure.
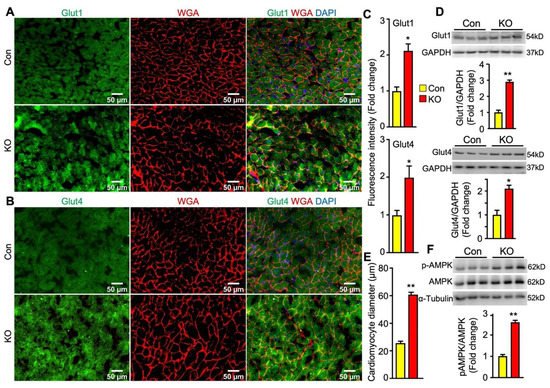
Figure 6. Adipose CGI-58 deficiency increases cardiomyocyte size and cardiac expression of proteins for Glut1, Glut4 and phosphorylated AMPK in chow-fed mice after cold stress. (A–C) Representative images and fluorescence intensity analysis of co-immunofluorescence staining of Glut1 or Glut 4 (green) with Alexa Fluor 594-conjugated WGA (red) in the cardiac tissues of the mice described under Figure 4. Quantification of fluorescence intensity was performed in three to five randomly selected microscopic fields per heart under 20x magnification (n = 4 mice/group). (D) Cardiac protein levels of Glut1 and Glut4 in the mice described under Figure 4. (E) The mean diameters of cardiomyocytes (µm) were measured from WGA-stained images in (A,B) with quantification of approximately 50–80 cells from 3 randomly selected image areas per heart under 20x magnification (n = 4 mice/group). (F) Levels of phosphorylated AMPK in the hearts of the mice described under Figure 4. * p < 0.05 and ** p < 0.001 vs. genotype by two-tailed unpaired Student’s t-test.
Energy fuel selection or switch has the potential to alter cellular energy status. AMP-activated protein kinase (AMPK) is the main energy sensor of cells [26]. In the cardiac tissue, activation of AMPK has been shown to promotes glucose uptake and glycolysis by recruiting Glut4 to the sarcolemmal membrane [27][28]. Interestingly, cardiac levels of phosphorylated AMPK were substantially increased in FAT-KO mice compared to the controls (Figure 6F). This finding suggests that AMPK activation may be a mechanistic link between adipose lipolysis inhibition and increased cardiac glucose utilization.
In contrary to glucose transporters, the fatty acid transporter CD36 protein level was significantly decreased in the hearts of FAT-KO mice compared to control mice (Figure 7). The downregulation in cardiac CD36 protein, together with the decrease in circulating FFA levels and the increase in cardiac glucose uptake, indicates that the hearts of adipose lipolysis-deficient mice heavily rely on glucose catabolism for energy.
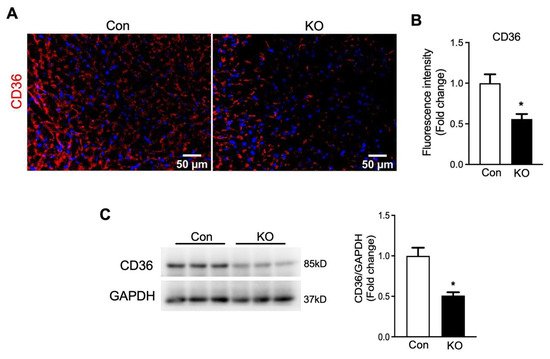
Figure 7. Deletion of adipose CGI-58 decreased cardiac CD36 protein level in chow-fed mice after cold stress. (A,B) Representative images and fluorescence intensity analysis of immunofluorescence staining of CD36 (red) with DAPI (blue) in the cardiac tissues of the mice described under Figure 4. Quantification of fluorescence intensity was performed in three to five randomly selected microscopic fields per heart under 20× magnification (n = 4 mice/group). (C) Cardiac protein levels of CD36 in the mice described under Figure 4. * p < 0.05 vs. genotype by two-tailed unpaired Student’s t-test.
References
- Cannon, B.; Nedergaard, J. Brown adipose tissue: Function and physiological significance. Physiol. Rev. 2004, 84, 277–359.
- Petrovic, N.; Walden, T.B.; Shabalina, I.G.; Timmons, J.A.; Cannon, B.; Nedergaard, J. Chronic peroxisome proliferator-activated receptor gamma (PPARgamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J. Biol. Chem. 2010, 285, 7153–7164.
- Wu, J.; Bostrom, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012, 150, 366–376.
- Virtanen, K.A.; Lidell, M.E.; Orava, J.; Heglind, M.; Westergren, R.; Niemi, T.; Taittonen, M.; Laine, J.; Savisto, N.J.; Enerback, S.; et al. Functional brown adipose tissue in healthy adults. N. Engl. J. Med. 2009, 360, 1518–1525.
- Van Marken Lichtenbelt, W.D.; Vanhommerig, J.W.; Smulders, N.M.; Drossaerts, J.M.; Kemerink, G.J.; Bouvy, N.D.; Schrauwen, P.; Teule, G.J. Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 2009, 360, 1500–1508.
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.H.; Doria, A.; et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517.
- Young, S.G.; Zechner, R. Biochemistry and pathophysiology of intravascular and intracellular lipolysis. Genes Dev. 2013, 27, 459–484.
- Lass, A.; Zimmermann, R.; Haemmerle, G.; Riederer, M.; Schoiswohl, G.; Schweiger, M.; Kienesberger, P.; Strauss, J.G.; Gorkiewicz, G.; Zechner, R. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell Metab. 2006, 3, 309–319.
- Yen, C.L.; Farese, R.V., Jr. Fat breakdown: A function for CGI-58 (ABHD5) provides a new piece of the puzzle. Cell Metab. 2006, 3, 305–307.
- Shin, H.; Ma, Y.; Chanturiya, T.; Cao, Q.; Wang, Y.; Kadegowda, A.K.G.; Jackson, R.; Rumore, D.; Xue, B.; Shi, H.; et al. Lipolysis in Brown Adipocytes Is Not Essential for Cold-Induced Thermogenesis in Mice. Cell Metab. 2017, 26, 764–777.e765.
- Shin, H.; Shi, H.; Xue, B.; Yu, L. What activates thermogenesis when lipid droplet lipolysis is absent in brown adipocytes? Adipocyte 2018, 27, 1–5.
- Schreiber, R.; Diwoky, C.; Schoiswohl, G.; Feiler, U.; Wongsiriroj, N.; Abdellatif, M.; Kolb, D.; Hoeks, J.; Kershaw, E.E.; Sedej, S.; et al. Cold-Induced Thermogenesis Depends on ATGL-Mediated Lipolysis in Cardiac Muscle, but Not Brown Adipose Tissue. Cell Metab. 2017, 26, 753–763.
- Thomou, T.; Mori, M.A.; Dreyfuss, J.M.; Konishi, M.; Sakaguchi, M.; Wolfrum, C.; Rao, T.N.; Winnay, J.N.; Garcia-Martin, R.; Grinspoon, S.K.; et al. Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature 2017, 542, 450–455.
- Nakamura, K.; Fuster, J.J.; Walsh, K. Adipokines: A link between obesity and cardiovascular disease. J. Cardiol. 2014, 63, 250–259.
- Akoumianakis, I.; Antoniades, C. The interplay between adipose tissue and the cardiovascular system: Is fat always bad? Cardiovasc. Res. 2017, 113, 999–1008.
- Bertero, E.; Maack, C. Metabolic remodelling in heart failure. Nat. Rev. Cardiol. 2018, 15, 457–470.
- Goldberg, I.J.; Trent, C.M.; Schulze, P.C. Lipid metabolism and toxicity in the heart. Cell Metab. 2012, 15, 805–812.
- Salatzki, J.; Foryst-Ludwig, A.; Bentele, K.; Blumrich, A.; Smeir, E.; Ban, Z.; Brix, S.; Grune, J.; Beyhoff, N.; Klopfleisch, R.; et al. Adipose tissue ATGL modifies the cardiac lipidome in pressure-overload-induced left ventricular failure. PLoS Genet. 2018, 14, e1007171.
- Foryst-Ludwig, A.; Kreissl, M.C.; Benz, V.; Brix, S.; Smeir, E.; Ban, Z.; Januszewicz, E.; Salatzki, J.; Grune, J.; Schwanstecher, A.K.; et al. Adipose Tissue Lipolysis Promotes Exercise-induced Cardiac Hypertrophy Involving the Lipokine C16:1n7-Palmitoleate. J. Biol. Chem. 2015, 290, 23603–23615.
- Ikaheimo, T.M. Cardiovascular diseases, cold exposure and exercise. Temperature 2018, 5, 123–146.
- Bordicchia, M.; Liu, D.; Amri, E.Z.; Ailhaud, G.; Dessi-Fulgheri, P.; Zhang, C.; Takahashi, N.; Sarzani, R.; Collins, S. Cardiac natriuretic peptides act via p38 MAPK to induce the brown fat thermogenic program in mouse and human adipocytes. J. Clin. Invest. 2012, 122, 1022–1036.
- Carper, D.; Coue, M.; Nascimento, E.B.M.; Barquissau, V.; Lagarde, D.; Pestourie, C.; Laurens, C.; Petit, J.V.; Soty, M.; Monbrun, L.; et al. Atrial Natriuretic Peptide Orchestrates a Coordinated Physiological Response to Fuel Non-shivering Thermogenesis. Cell Rep. 2020, 32, 108075.
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 1, 785–789.
- Sergeeva, I.A.; Christoffels, V.M. Regulation of expression of atrial and brain natriuretic peptide, biomarkers for heart development and disease. Biochim. Biophys Acta 2013, 1832, 2403–2413.
- Pessin, J.E.; Bell, G.I. Mammalian facilitative glucose transporter family: Structure and molecular regulation. Annu. Rev. Physiol. 1992, 54, 911–930.
- Kahn, B.B.; Alquier, T.; Carling, D.; Hardie, D.G. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005, 1, 15–25.
- Till, M.; Ouwens, D.M.; Kessler, A.; Eckel, J. Molecular mechanisms of contraction-regulated cardiac glucose transport. Biochem. J. 2000, 346, 841–847.
- Russell, R.R., III; Bergeron, R.; Shulman, G.I.; Young, L.H. Translocation of myocardial GLUT-4 and increased glucose uptake through activation of AMPK by AICAR. Am. J. Physiol. 1999, 277, H643–H649.
More
Information
Subjects:
Endocrinology & Metabolism
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
852
Revisions:
2 times
(View History)
Update Date:
09 Feb 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No