 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kenji Miyado | + 2118 word(s) | 2118 | 2021-12-08 09:43:40 | | | |
| 2 | Woojin Kang | + 3 word(s) | 2121 | 2021-12-09 05:30:28 | | |
Video Upload Options
The tricarboxylic acid (TCA) cycle, also known as the citrate acid cycle, is a series of chemical reactions to form energy required for cellular function through the oxidation of acetyl-CoA derived from carbohydrates, fats, and proteins. There are eight enzymes in the TCA cycle that oxidize acetyl-coenzyme A (acetyl-CoA), and genetic or non-genetic alterations in these enzymes are closely associated with human diseases, especially cancer and neurodegeneration, but the role of these eight enzymes remains unclear.
1. Background
The tricarboxylic acid (TCA) cycle is the main source of cellular energy and participates in many metabolic pathways in cells. Recent reports indicate that dysfunction of TCA cycle-related enzymes causes human diseases, such as neurometabolic disorders and tumors, have attracted increasing interest in their unexplained roles. The diseases which develop as a consequence of loss or dysfunction of TCA cycle-related enzymes are distinct, suggesting that each enzyme has a unique function.
2. TCA-Related Enzymes and Diseases Arising from Their Dysfunction
| Enzymes | Abbreviation | Localization | Diseases | References |
|---|---|---|---|---|
| Citrate synthase | CS | Mitochondria | Cell death (in a human cell line, in vitro) | [2] |
| Citrate lyase | ACLY | Cytosol | Atherosclerotic plaques (in mice, in vivo) | [4] |
| Extra-mitochondrial citrate synthase | eCS | Cytosol | Decrease in age-dependent male fertility (in mice, in vivo) |
[20] |
| Aconitase | ACO1 | Cytosol | Encephalopathy (in humans, in vivo) | [3][6][7] |
| ACO2 | Mitochondria | Optic atrophy (in humans, in vivo) | ||
| Isocitrate dehydrogenase | IDH1 | Cytosol | Gliomas, acute myeloid leukemia (in humans, in vivo) |
[8][3][10] |
| IDH2 | Mitochondria | |||
| Succinate dehydrogenase | SDHA | Mitochondria | Paragangliomas (in humans, in vivo) | [3][13] |
| SDHB | Gastrointestinal stromal tumors, paragangliomas, renal cell carcinoma, T-cell acute leukemia (in humans, in vivo) |
[3][12][14][15][16] | ||
| SDHC | Gastrointestinal stromal tumors, paragangliomas (in humans, in vivo) |
[3][12][16] | ||
| SDHD | Gastrointestinal stromal tumors, paragangliomas (in humans, in vivo) |
[3][12][16] | ||
| Fumarase (fumarase hydratase) |
FH | Mitochondria | Encephalopathy, leiomyomas, leiomyomatosis, renal cell cancer, ovary cystadenomas, breast cancer (in humans, in vivo) | [3][18] |
| Cytosol | ||||
| α-ketoglutarate dehydrogenase | OGDH | Mitochondria | Neurological disorder (in humans, in vivo) | [21] |
| Malate dehydrogenase | MDH1 | Cytosol | Encephalopathy (in a human cell line, in vitro) | [22][23] |
| MDH2 | Mitochondria | |||
| Malic enzyme | ME1 | Cytosol | Unknown | [24][25] |
| ME2 | Mitochondria | Idiopathic generalized epilepsy (in humans, in vivo) | [24][26] | |
| ME3 | Mitochondria | Unknown | [24] | |
| Glutamate-oxaloacetate transaminase | GOT1 | Cytosol | Unknown | [27] |
| GOT2 | Mitochondria | Neurometabolic disorder (in humans, in vivo) | [28] |
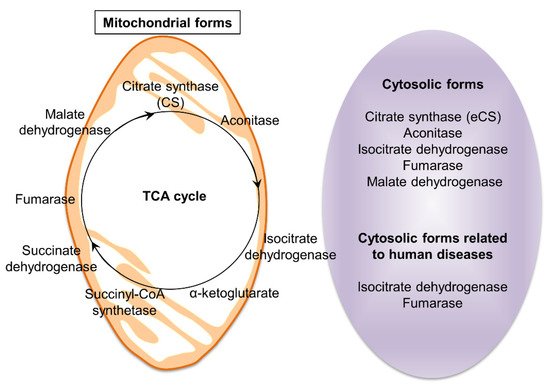
3. Predicted Existence of Extra-Mitochondrial TCA (eTCA) Cycle
References
- Verstraete, K.; Verschueren, K.H.; Dansercoer, A.; Savvides, S.N. Acetyl-CoA is produced by the citrate synthase homology module of ATP-citrate lyase. Nat. Struct. Mol. Biol. 2021, 28, 636–638.
- Cai, Q.; Zhao, M.; Liu, X.; Wang, X.; Nie, Y.; Li, P.; Liu, T.; Ge, R.; Han, F. Reduced expression of citrate synthase leads to excessive superoxide formation and cell apoptosis. Biochem. Biophys. Res. Commun. 2017, 485, 388–394.
- Raimundo, N.; Baysal, B.E.; Shadel, G.S. Revisiting the TCA cycle: Signaling to tumor formation. Trends Mol. Med. 2011, 17, 641–649.
- Baardman, J.; Verberk, S.G.; Van der Velden, S.; Gijbels, M.J.; van Roomen, C.P.; Sluimer, J.C.; Broos, J.Y.; Griffith, G.R.; Prange, K.H.; van Weeghel, M. Macrophage ATP citrate lyase deficiency stabilizes atherosclerotic plaques. Nat. Commun. 2020, 11, 1–15.
- Khodagholi, F.; Shaerzadeh, F.; Montazeri, F. Mitochondrial aconitase in neurodegenerative disorders: Role of a metabolism-related molecule in neurodegeneration. Curr. Drug Targets 2018, 19, 973–985.
- Abela, L.; Spiegel, R.; Crowther, L.M.; Klein, A.; Steindl, K.; Papuc, S.M.; Joset, P.; Zehavi, Y.; Rauch, A.; Plecko, B. Plasma metabolomics reveals a diagnostic metabolic fingerprint for mitochondrial aconitase (ACO2) deficiency. PLoS ONE 2017, 12, e0176363.
- Chen, Y.; Cai, G.H.; Xia, B.; Wang, X.; Zhang, C.C.; Xie, B.C.; Shi, X.C.; Liu, H.; Lu, J.F.; Zhang, R.X. Mitochondrial aconitase controls adipogenesis through mediation of cellular ATP production. FASEB J. 2020, 34, 6688–6702.
- Martinez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102.
- Martínez-Reyes, I.; Diebold, L.P.; Kong, H.; Schieber, M.; Huang, H.; Hensley, C.T.; Mehta, M.M.; Wang, T.; Santos, J.H.; Woychik, R. TCA cycle and mitochondrial membrane potential are necessary for diverse biological functions. Mol. Cell 2016, 61, 199–209.
- Uckermann, O.; Juratli, T.A.; Galli, R.; Conde, M.; Wiedemuth, R.; Krex, D.; Geiger, K.; Temme, A.; Schackert, G.; Koch, E.; et al. Optical analysis of glioma: Fourier-transform infrared spectroscopy reveals the IDH1 mutation status. Clin. Cancer Res. 2018, 24, 2530–2538.
- Hoekstra, A.S.; Bayley, J.-P. The role of complex II in disease. Biochim. Biophys. Acta (BBA)-Bioenerg. 2013, 1827, 543–551.
- Astuti, D.; Hart-Holden, N.; Latif, F.; Lalloo, F.; Black, G.C.; Lim, C.; Moran, A.; Grossman, A.B.; Hodgson, S.V.; Freemont, A. Genetic analysis of mitochondrial complex II subunits SDHD, SDHB and SDHC in paraganglioma and phaeochromocytoma susceptibility. Clin. Endocrinol. 2003, 59, 728–733.
- Tufton, N.; Ghelani, R.; Srirangalingam, U.; Kumar, A.V.; Drake, W.M.; Iacovazzo, D.; Skordilis, K.; Berney, D.; Khoo, B.; Akker, S.A. SDHA mutated paragangliomas may be at high risk of metastasis. Endocr. Relat. Cancer 2017, 24, L43–L49.
- Vanharanta, S.; Buchta, M.; McWhinney, S.R.; Virta, S.K.; Peçzkowska, M.; Morrison, C.D.; Lehtonen, R.; Januszewicz, A.; Järvinen, H.; Juhola, M. Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. Am. J. Hum. Genet. 2004, 74, 153–159.
- Baysal, B.E. A recurrent stop-codon mutation in succinate dehydrogenase subunit B gene in normal peripheral blood and childhood T-cell acute leukemia. PLoS ONE 2007, 2, e436.
- Stratakis, C.; Carney, J. The triad of paragangliomas, gastric stromal tumours and pulmonary chondromas (Carney triad), and the dyad of paragangliomas and gastric stromal sarcomas (Carney–Stratakis syndrome): Molecular genetics and clinical implications. J. Intern. Med. 2009, 266, 43–52.
- Scheffler, I.E. Mitochondria; John Wiley & Sons: Hoboken, NJ, USA, 2011.
- Crooks, D.R.; Maio, N.; Lang, M.; Ricketts, C.J.; Vocke, C.D.; Gurram, S.; Turan, S.; Kim, Y.Y.; Cawthon, G.M.; Sohelian, F.; et al. Mitochondrial DNA alterations underlie an irreversible shift to aerobic glycolysis in fumarate hydratase-deficient renal cancer. Sci Signal. 2021, 14, eabc4436.
- Raimundo, N.; Ahtinen, J.; Fumić, K.; Barić, I.; Remes, A.M.; Renkonen, R.; Lapatto, R.; Suomalainen, A. Differential metabolic consequences of fumarate hydratase and respiratory chain defects. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2008, 1782, 287–294.
- Kang, W.; Harada, Y.; Yamatoya, K.; Kawano, N.; Kanai, S.; Miyamoto, Y.; Nakamura, A.; Miyado, M.; Hayashi, Y.; Kuroki, Y.; et al. Extra-mitochondrial citrate synthase initiates calcium oscillation and suppresses age-dependent sperm dysfunction. Lab. Investig. 2020, 100, 583–595.
- Gibson, G.E.; Park, L.C.; Sheu, K.-F.R.; Blass, J.P.; Calingasan, N.Y. The α-ketoglutarate dehydrogenase complex in neurodegeneration. Neurochem. Int. 2000, 36, 97–112.
- Broeks, M.H.; Shamseldin, H.E.; Alhashem, A.; Hashem, M.; Abdulwahab, F.; Alshedi, T.; Alobaid, I.; Zwartkruis, F.; Westland, D.; Fuchs, S.; et al. MDH1 deficiency is a metabolic disorder of the malate-aspartate shuttle associated with early onset severe encephalopathy. Hum. Genet. 2019, 138, 1247–1257.
- Ait-El-Mkadem, S.; Dayem-Quere, M.; Gusic, M.; Chaussenot, A.; Bannwarth, S.; François, B.; Genin, E.C.; Fragaki, K.; Volker-Touw, C.L.; Vasnier, C. Mutations in MDH2, encoding a Krebs cycle enzyme, cause early-onset severe encephalopathy. Am. J. Hum. Genet. 2017, 100, 151–159.
- Hassinen, I.E. Signaling and regulation through the NAD+ and NADP+ networks. Antioxid. Redox Signal. 2019, 30, 857–874.
- Lu, Y.-X.; Ju, H.-Q.; Liu, Z.-X.; Chen, D.-L.; Wang, Y.; Zhao, Q.; Wu, Q.-N.; Zeng, Z.-l.; Qiu, H.-B.; Hu, P.-S. ME1 regulates NADPH homeostasis to promote gastric cancer growth and metastasis. Cancer Res. 2018, 78, 1972–1985.
- Lenzen, K.P.; Heils, A.; Lorenz, S.; Hempelmann, A.; Sander, T. Association analysis of malic enzyme 2 gene polymorphisms with idiopathic generalized epilepsy. Epilepsia 2005, 46, 1637–1641.
- Kremer, D.M.; Nelson, B.S.; Lin, L.; Yarosz, E.L.; Halbrook, C.J.; Kerk, S.A.; Sajjakulnukit, P.; Myers, A.; Thurston, G.; Hou, S.W. GOT1 inhibition promotes pancreatic cancer cell death by ferroptosis. Nat. Commun. 2021, 12, 1–13.
- Ramos, R.J.; van Karnebeek, C.D.; Ciapaite, J.; Pras-Raves, M.; Waterham, H.R.; Wanders, R.J.; Jans, J.J.; Verhoeven-Duif, N.M. Metabolic Consequences of GOT2 Deficiency. New Insight into Vitamin B6 Metabolism and Related Diseases. Ph.D. Thesis, Utrecht University, Utrecht, The Netherlands, 2019.
- Murai, S.; Ando, A.; Ebara, S.; Hirayama, M.; Satomi, Y.; Hara, T. Inhibition of malic enzyme 1 disrupts cellular metabolism and leads to vulnerability in cancer cells in glucose-restricted conditions. Oncogenesis 2017, 6, e329.
- Mellis, A.-T.; Misko, A.L.; Arjune, S.; Liang, Y.; Erdélyi, K.; Ditrói, T.; Kaczmarek, A.T.; Nagy, P.; Schwarz, G. The role of glutamate oxaloacetate transaminases in sulfite biosynthesis and H2S metabolism. Redox Biol. 2021, 38, 101800.
- Cardaci, S.; Ciriolo, M.R. TCA cycle defects and cancer: When metabolism tunes redox state. Int. J. Cell Biol. 2012, 2012, 161837.
- Wachnowsky, C.; Hendricks, A.L.; Wesley, N.A.; Ferguson, C.; Fidai, I.; Cowan, J.A. Understanding the mechanism of cluster assembly on eukaryotic mitochondrial and cytosolic aconitase. Inorg. Chem. 2019, 58, 13686–13695.
- Johnson, N.B.; Deck, K.M.; Nizzi, C.P.; Eisenstein, R.S. A synergistic role of IRP1 and FBXL5 proteins in coordinating iron metabolism during cell proliferation. J. Biol. Chem. 2017, 292, 15976–15989.
- Leshets, M.; Silas, Y.B.H.; Lehming, N.; Pines, O. Fumarase: From the TCA cycle to DNA damage response and tumor suppression. Front. Mol. Biosci. 2018, 5, 68.
- Eprintsev, A.T.; Fedorin, D.N.; Starinina, E.V.; Igamberdiev, A.U. Expression and properties of the mitochondrial and cytosolic forms of fumarase in germinating maize seeds. Physiol. Plant. 2014, 152, 231–240.
- Dyson, B.C.; Miller, M.A.; Feil, R.; Rattray, N.; Bowsher, C.G.; Goodacre, R.; Lunn, J.E.; Johnson, G.N. FUM2, a cytosolic fumarase, is essential for acclimation to low temperature in Arabidopsis thaliana. Plant. Physiol. 2016, 172, 118–127.
- Himpsl, S.D.; Shea, A.E.; Zora, J.; Stocki, J.A.; Foreman, D.; Alteri, C.J.; Mobley, H.L.T. The oxidative fumarase FumC is a key contributor for E. coli fitness under iron-limitation and during UTI. PLoS Pathog. 2020, 16, e1008382.
- Yogev, O.; Yogev, O.; Singer, E.; Shaulian, E.; Goldberg, M.; Fox, T.D.; Pines, O. Fumarase: A mitochondrial metabolic enzyme and a cytosolic/nuclear component of the DNA damage response. PLoS Biol. 2010, 8, e1000328.
- Van Lith, S.A.; Navis, A.C.; Lenting, K.; Verrijp, K.; Schepens, J.T.; Hendriks, W.J.; Schubert, N.A.; Venselaar, H.; Wevers, R.A.; van Rooij, A.; et al. Identification of a novel inactivating mutation in Isocitrate Dehydrogenase 1 (IDH1-R314C) in a high grade astrocytoma. Sci. Rep. 2016, 6, 30486.
- Meemongkolkiat, T.; Allison, J.; Seebacher, F.; Lim, J.; Chanchao, C.; Oldroyd, B.P. Thermal adaptation in the honeybee (Apis mellifera) via changes to the structure of malate dehydrogenase. J. Exp. Biol. 2020, 223, jeb228239.
- López-Lázaro, M. The warburg effect: Why and how do cancer cells activate glycolysis in the presence of oxygen? Anti Cancer Agents Med. Chem. Former. Curr. Med. Chem. Anti Cancer Agents 2008, 8, 305–312.
- Liberti, M.V.; Locasale, J.W. The Warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218.
- Panfoli, I.; Calzia, D.; Ravera, S.; Bruschi, M.; Tacchetti, C.; Candiani, S.; Morelli, A.; Candiano, G. Extramitochondrial tricarboxylic acid cycle in retinal rod outer segments. Biochimie 2011, 93, 1565–1575.

