 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Elba Mauriz | + 3157 word(s) | 3157 | 2021-11-29 10:44:30 | | | |
| 2 | Amina Yu | + 32 word(s) | 3189 | 2021-12-06 05:10:06 | | |
Video Upload Options
This work compiles recent advancements on the discovery of SARS-CoV2 inhibitors by SPR analysis. One of the major benefits of SPR biosensing is the possibility of easily investigating molecular interactions. Owing to this quality, SPR biosensor technology has become a first-line tool for analyzing the binding properties of potential drug candidates against COVID-19. Overall, SPR biosensors seem to offer a single methodology for attaining full kinetic profiles while providing the screening of fragment libraries and the validation of fragment hits.
1. Introduction
Past pandemics of severe acute respiratory syndrome (SARS-CoV-1) in 2003, influenza (H1N1, 1918 and 2009; H2N2, 1958; H3N2, 1968), Middle East respiratory syndrome (MERS) in 2013, and Ebola virus disease in 2014, had previously menaced global health. However, the immense impact of the COVID-19 pandemic outburst in the health condition and socioeconomic prospects of the world population has reached unknown proportions [1]. The unexpected risks of recurrent virus outbreaks have stressed the need for searching efficient strategies to prevent viral infections [2]. Generally, antiviral therapeutic interventions rely on the development of candidate drugs following the ‘one bug, one drug’ approach [2][3]. This paradigm is insufficient to lead the fight against viruses due to: (i) the increasing number of emerging viruses; (ii) the presence of undiscovered mammalian virus in the wildlife reservoir; (iii) the lack of broad-spectrum antiviral agents; and (iv) the appearance of drug resistances [4].
Thus, the amelioration of viral infections requires the enlargement of drug candidates with potent antiviral effect and broad-spectrum activities [3]. Novel drug discovery strategies rely on the high throughput screening of different classes of antiviral agents [5]. For example, the increase of antibody drug candidates has provided an efficient approach to treat viral infections due to their specificity [6][7]. However, this selectivity is directed against specific antigens thus limiting their spectrum of activity. To overcome this problem, the design of antiviral agents has concentrated on the screening of low-molecular-weight fragments that can bind to membrane protein drug targets [5][8]. Fragment-based drug discovery has enabled the identification of a significant number of drugs approved for clinical use [9]. Owing to their low molecular mass, the binding with their target of interest may result in low affinity interactions that require the employment of highly sensitive biophysical techniques for their estimation [8]. Among them, nuclear magnetic resonance (NMR) [10], X-ray crystallography [11], mass spectroscopy [12], isothermal titration calorimetry [13], protein thermal shift [14], affinity capillary electrophoresis [15], weak affinity chromatography [16], have been commonly used to assess protein–drug interactions. More recently, label-free biosensors based on Optical Waveguide Grating (OWG) [17] and reflectometric interference spectroscopy (RIfS) [18] have emerged as valuable tools for evaluating biomolecular interactions [8].
From this perspective, surface plasmon resonance (SPR) biosensors seem to be ideally suited to address this challenge. The application of SPR biosensor technology in the pharmaceutical field has been extensively employed to investigate the binding properties of a broad range of drug candidates from nucleic acids to antibodies [19]. SPR biosensors provide label-free evaluations of molecular interactions between viral targets and their specific recognition element immobilized on the biosensor surface [8][20]. Specifically, SPR sensor devices detect the refraction index variations resulting from the interaction between the analyte and the immobilized bioreceptor occurring on the sensing layer after recognizing the target analyte ( Figure 1 ) [21][22]. The main advantages of SPR biosensor over others biophysical techniques rely on both the short-time response and the capacity to monitor binding activities in real time. SPR analysis offers precise data on the kinetics and thermodynamics of the binding between virus target and the immobilized bioreceptor. SPR sensorgrams provide the association, equilibrium and dissociation phases observed after the injection of the analyte onto the immobilized bioreceptor surface [5][23]. By monitoring binding interactions, SPR biosensors inform on the association (Ka) and dissociation rates (Kd), equilibrium dissociation constants (KD) and stoichiometry of the interacting biomolecules depending on their binding affinity [6][8][24]. The kinetic information supplied by SPR biosensors is particularly useful for the rapid screening of drug candidates such as virus entry inhibitors. To date, SPR strategies have been successfully applied to the label-free sensitive detection of a wide variety of biomolecules including G-protein coupled receptors, antibodies, aptamers and enzymes [8][24][25].
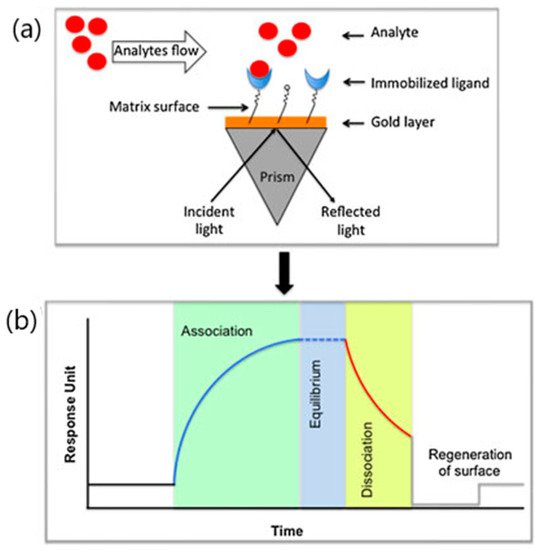
The role of SPR biosensors in pharmaceutical analysis and viral diagnosis has been previously explored [5][8][19][20][26]. However, the specific utility of SPR biosensors for high-throughput screening of viral inhibitors in the current pandemic context has not been sufficiently addressed [5][6]. Therefore, the aim of this work is to compile recent developments of SPR biosensor approaches in targeting SARS-CoV-2 entry into host cells. This review specifically concentrates on the identification and prioritization of viral inhibitors through the characterization of their binding and kinetic parameters. Current research in SPR antiviral drug development involving fragment-based drug discovery screening of COVID-19 candidate drugs is also reviewed.
2. Viral Entry and Drug Screening
Viral entry is an indispensable condition to initiate the infection cycle by releasing the viral genome into the infected cell [2][5][27]. The entry process in enveloped viruses involves first the specific binding of the viral surface proteins to the receptors of the cell to be infected and the subsequent viral fusion with the host cell membrane [2][5][27]. Depending on the virus class, the activation of one type or a set of viral envelope glycoproteins, triggers conformational changes in the membrane proteins of both, the virus and the host [4]. A series of intermediate stages comprising pre-fusion, pre-hairpin and post-fusion membrane structural changes complete the viral fusion into the host cell ( Figure 2 ).

Despite the process complexity, the vast majority of viruses present a similar cell entry route regardless of their morphology, genome structure and life cycle. This feature is especially interesting for the design of universal drug strategies that contain viral infections. Tackling viral entry guarantees easier access to the virus target sites while preserving the integrity of the host cell [5]. Particularly, blocking the entry process into host cells seems to be a promising way to protect against viral infection [2]. Among viral entry inhibitors, the most common approaches for inhibiting virus fusion to host cells are based on peptides and small molecules. Although peptide-based inhibitors have been extensively used to block viral fusion, recent strategies have set the focus on the screening of low-molecular-weight compounds known as fragments [8][28].
Fragment screening has led to the discovery of a considerable number, more than 30 clinical drug candidates in the last 20 years [24]. The utilization of fragment-derived compounds has produced significant benefits in drug discovery since it provides large areas of chemical space [29] and lesser probability of undesired interaction. Additionally, the optimization of the molecular size may lead to improve the affinity and selectivity of the fragments. However, binding between low molecular weight compounds and their ligands often results in low-affinity interactions. Therefore, detection of low fragments requires the employment of highly sensitive biophysical methods (NMR, X-ray crystallography, spectrometry, and so on).
In this context, the SPR biosensor approach has contributed to the characterization and pharmacokinetic profiling of a variety of fragment-based virus inhibitors in the last decade. The role of SPR biosensors is not only limited to complementing other screening techniques but also to informing on the kinetics and binding constants of the low-affinity fragments. Recent advances in SPR biosensor technology have permitted the rapid screening, identification, validation and confirmation of fragments directed against different types of targets (proteases) [30], kinases [29], GPCRs [8] and protein–protein interactions (PPIs) [31]. The following sections describe experimental considerations as well as strengths and limitations in the detection of virus entry inhibitors against SARS-CoV-2.
3. SPR Analysis of SARS-CoV-2 Entry Inhibitors
Several SPR approaches have been involved in both pharmaceutical profiling of SARS-CoV-2 entry inhibitors and characterization of the kinetics during the entry process of SARS-CoV-2 into host cells. Most strategies focus on the ability of drug candidates to block ACE2-S-RBD interactions by examining their binding affinities with both ACE2 and RBD proteins. However, a limited number of studies have come forward to assume the detection of affinity constants while screening potential viral inhibitors. Table 1 summarizes the most singular approaches involving either the kinetic analysis or the screening of SARS-CoV-2 inhibitors capable of blocking the ACE2 receptor. The analytical performance characteristics of each method are specifically discussed below.
| Target Analyte (MW, Common Uses) | Analytical Approach | SPR Instrument | Binding Affinities (KD: Equilibrium Dissociation Constant) | Reference |
|---|---|---|---|---|
| Demethylzeylasteral (480.59 (g/mol); immunosuppressor, anti-inflammatory, anti-tumoral) | Protein–protein interaction testing Screening and kinetic analysis (binding to ACE2 and S-RBD) Competition assay (blocking RBD-ACE2 interactions) |
Biacore T200 (Washington, DC, USA) or S200 instrument (GE Healthcare Life Sciences). | KD = 1.736 mM; kon (1/Ms) = 1989; koff (×10−3 1/s) = 3.345 (ACE2) KD = 1.039 mM (S-RBD) |
[32] |
| Sodium lifitegrast (among 21 screened compounds) (637.5 g/mol; keratoconjunctivitis) | SPR screening combined approach Kinetic analysis (binding to S-RBD) Competition assay (blocking RBD-ACE2 interactions) |
Biacore S200 system (GE Healthcare Life Sciences) |
KD = 1.92 nM (sodium lifitegrast) (KD < 3 mM: rest of compounds) (kon and koff values N/A) | [33] |
| Thymoquinone (164,201 g/mol; antioxidant, anti-inflammatory, chemotherapic) | Binding affinity to ACE2 receptors | Biacore T200 System (GE Healthcare, Uppsala, Sweden) |
KD = 32.140 mM (kon and koff values N/A) | [34] |
| Ginsenoside Ra2, ginsenoside Rb1, ginsenoside Rb3, glycyrrhizic acid and berberine chloride (1211.38, 1109.29, 1079.27, 822.94 and 371.81 g/mol respectively; herbs within clinically effective TCM schemes) |
Binding activity of TCM-derived components with SARS-CoV-2 S1 subunit | BIAcore T200 instrument (BIAcore T200, GE Healthcare, Chicago, IL, USA) |
KD = 55.6 mM (ginsenoside Ra2); KD = 29.7 mM (ginsenoside Rb3); KD = 2.0 mM (ginsenoside Rb1); KD = 66.8 mM (glycyrrhizic acid); KD = 23.9 mM (berberine chloride) (kon and koff values N/A) |
[35] |
| Quinoline-2-carboxylic acids (3 compounds) (173.168 g/mol, Ephedra sinica extracts; lung diseases treatment) | Binding affinity constants (to S-RBD) Competition assay (blocking RBD-ACE2 interactions) |
BIAcore T200 instrument (GE Healthcare Life Sciences) | KD = 0.60–5.37 mM (kon and koff values N/A) |
[36] |
| Radix Scutellariae extract: Oroxylin A (284.26 g/mol; respiratory diseases, diabetes, diarrhea treatment) | Binding affinity constants (to ACE2) | Open SPR™ (Nicoya Lifesciences, Waterloo, Canada) | KD = 9.72 × 10−6 M (kon and koff values N/A) |
[37] |
| Polyphenols: corilagin and TGG (636.46 and 423.40 g/mol, respectively; antioxidant, anti-inflammatory, and antidiabetic treatments) | Binding affinity constants (to S-RBD) Binding affinity of TGG and corilagin with RBD mutations of three main SARS-CoV-2 variants Competition assay (blocking RBD–ACE2 interactions) |
Biacore T200 instrument (GE Healthcare) |
KD = 1.8 nM (corilagin) KD = 1.3 nM (TGG) (kon and koff values N/A) |
[38] |
| Erythrodiol (442.7 g/mol; Momordica charantia component: treatment of nephropathy, neuropathy, gastroparesis, cataracts and atherosclerosis) |
Binding affinity constants (to SARS-CoV-2 S1 subunit) | Open SPR instrument (Nicoya Lifescience, ON, Canada) |
KD = 1.15 μM (kon and koff values N/A) |
[39] |
| Histamine H1 receptor antagonists: doxepin, chlorpheniramine, and doxylamine (279.376, 274.788 and 270.369 g/mol respectively; allergic rhinitis, allergic conjunctivitis and allergic dermatitis) | Analysis of bimolecular interactions with ACE2 receptor | Open SPRTM (Nicoya, Waterloo, Canada) | KD = 9.54 mM (doxepin); KD = 0.30 mM (chlorpheniramine); KD = 47.3 mM (doxylamine) (kon and koff values N/A) |
[40] |
| Azelastine (381,898 g/mol; allergic conjunctivitis) | Kinetic analysis (binding to ACE2 receptor) | Open SPRTM (Nicoya, Waterloo, Canada) | KD = 0.258 μM (kon and koff values N/A) |
[41] |
| Desloratadine and loratadine (310.82 and 382.88 g/mol; treatment of allergic disease) | Kinetic analysis (binding to ACE2 receptor) | Open SPRTM (Nicoya, Waterloo, Canada) | KD = 9.13 μM (desloratadine); KD = 0.1.02 μM (loratadine) (kon and koff values N/A) |
[42] |
| Chloroquine (CQ) and hydroxychloroquine (HCQ) (319.872 and 335.872 g/mol; antimalarial drugs) |
Kinetic analysis (binding to ACE2 receptor) | Open SPRTM (Nicoya, Waterloo, Canada) | KD = (7.31 ± 0.62) × 10−7 M (CQ) KD = (4.82 ± 0.87) × 10−7 M (HCQ) (kon and koff values N/A) |
[43] |
| Trifluoperazine (Tri); thioridazine (Thi); chlorpromazine (Chl), aripiprazole (Ari), tiapride (Tia) (407.497, 370.6, 318.86, 448.385 and 328,427 g/mol, respectively; antipsychotic drugs) | Kinetic analysis (binding to ACE2 receptor) | Open SPRTM (Nicoya, Waterloo, Canada) | KD = (7.03 ± 3.28) × 10−6 M (Tri); KD = (8.91 ± 5.25) × 10−5 M (Thi); KD = (1.38 ± 0.38) × 10−5 M (Chl); KD = (7.88 ± 0.49) × 10−6 M (Ari), and (3.33 ± 3.13) × 10−5 M (Tia) (kon and koff values N/A) |
[44] |
MW: Molecular weight; kon = association rate; koff = dissociation rate; KD = equilibrium dissociation constants; N/A = not available; TCM: traditional Chinese medicine.
Zhu et al. [32] investigated protein–protein interactions between human ACE2 and SARS-CoV-2 RBD proteins in search of drug inhibitors. The targeting of surface interactions allowed the discovery of demethylzeylasteral among a library of 960 compounds. First, the binding ability of the proteins was evaluated using ACE2-His as the captured ligand on the SPR sensor chip and S-RBD-mFc as the analyte. To improve the screening efficiency, several models established the contact and dissociation times according to the molecular weights of both the target protein and the screened compound. The screening and affinity analysis of low-molecular-weight compounds was performed following three steps, namely: clean screen, binding level screen and affinity screen. Clean screen permitted the exclusion of 13 compounds more likely to bind residually on the sensor chip surface due to their viscosity. Subsequently, binding level screen enabled the identification of compounds that could bind to the targets of interest and finally, a dose–response curve was assayed to determine their binding affinity. Since residence time was crucial for evaluating the drug efficacy, the association rate constant (k on) and the duration of target occupancy measured by the dissociation rate constant (k off) were examined through a kinetic fitting model. The results confirmed by Isothermal Titration Calorimetry (ITC), showed that demethylzeylasteral could bind to ACE2 and S-RBD with KD values of 1.736 and 1.039 M, respectively. The capacity for blocking ACE2-S-RBD interactions in a dose-dependent manner was also demonstrated by demethylzeylasteral via an SPR-based competitive assay ( Figure 3 ). These results were confirmed by virtual docking, thereby showing that demethylzeylasteral could dock into the ACE2 binding area of S-RBD protein. Therefore, the feasibility of SPR technology as the main method to screen drug inhibitors without the support of traditional screening methods was demonstrated. Moreover, the utility of SPR analysis as a primary screening method has paved the way to acquire information on kinetics and affinity constants more easily by using a single-step format.
In this framework, the antiviral effect of histamine H1 antagonist approved drugs loratadine (LOR) and desloratadine (DES) has also been investigated. A combined approach involving cell membrane chromatography and SPR sensing was developed to determine the binding affinity of loratadine and desloratadine to ACE2 receptor [42]. On the one hand, CMC showed that the specific binding durations of LOR and DES to ACE2 receptor were 2.49 and 51.8 min, respectively, showing that desloratadine could bind to ACE2 receptor more strongly than loratadine. These findings were confirmed by SPR assays where desloratadine exhibited higher binding constants (KD = 9.13 ± 0.67 × 10 −6 M) than loratadine (1.02 ± 0.38 × 10 −7 M). Similarly, molecular docking determinations proved that desloratadine could bind to ACE2 through one hydrogen bond with LYS31 while loratadine did not form hydrogen bonds, thus validating the SPR results. Since the blocking effect was only measured on the entry of SARS-CoV-2 spike pseudotyped virus into ACE2h cells, the efficacy of loratadine and desloratadine may need further evaluation in native viruses and animal models.
A similar approach made use of phenolic compounds to test their anti-SARS-CoV-2 Mpro activity [45]. This entry offers an interesting procedure comprising the combination of enzyme inhibition, SPR-, and docking-based assays to evaluate the inhibition capacity of dietary tannins (ellagitannins (punicalagin: PA and ellagic acid: EA) and gallotannins (tannic acid: TA, pentagalloyl glucose: PGG, ginnalin A and gallic acid: GA), and their gut microbial metabolites, urolithins UB and pyrogallol PYG. The optimization of the SPR conditions include the analysis of binding parameters, flow rate and regeneration buffers. Ellagitannins PA and EA showed high binding affinity to the SARS-CoV-2 Mpro protein with KD values of 6.8 × 10 −6 and 2.7 × 10 −6 M, respectively. Similarly, gallotannins TA, PGG and GA also exhibited a strong binding capacity with high KD values of 1.13, 4.33 and 1.18 × 10 −6 M, respectively. Lastly, KD of UB and PYG were 5.27 × 10 −5 and 3.59 × 10 −6 M, respectively. Although SPR experiments could not predict the specific binding to the catalytic or allosteric domain to the protein, this work demonstrated that biochemical assays, biophysical-based binding assays, and computational approach can be combined successfully for evaluating the inhibition and binding affinities of drug candidates.
4. Conclusions and Future Perspectives
This work compiles recent advancements on the discovery of SARS-CoV2 inhibitors by SPR analysis. One of the major benefits of SPR biosensing is the possibility of easily investigating molecular interactions. Owing to this quality, SPR biosensor technology has become a first-line tool for analyzing the binding properties of potential drug candidates against COVID-19.
Primarily, SPR biosensors have been used to measure the binding affinities between an immobilized protein and the selected compound with antiviral properties. Most of the SPR approaches follow this route to characterize the binding specificity between interactants. Recent progress in SPR technology has enabled the detection of weak interactions occurring between small molecules and immobilized proteins with equilibrium dissociation constants in the mM range. In general, SPR-based assays make use of the immobilization of either ACE2 receptor expressed on the membrane of host cells or the main protease Mpro. By utilizing this detection scheme, SPR biosensors are capable of generating a full kinetic profile of the interaction with SARS-CoV2 entry inhibitors. When monitoring drug-ACE2 binding constants, SPR experiments usually focused on the capacity of the antiviral compound to block the interaction with the RBD of the virus spike protein. Alternatively, Mpro-based immobilization formats addressed drug discovery from the perspective of the inhibitory compound ability to prevent virus replication and disrupt the virus life cycle.
In both cases, target molecules subjected to SPR analysis commonly come from natural origin, mainly fruits and vegetables, or pharmaceutical drug synthesis. Among the latter, commercially approved drugs are normally selected for drug repurposing studies according to previously reported therapeutic uses. Thus, a broad range of compounds, including antiparasitic, antibiotics or antipsychotic drugs, have been repositioned as possible anti-COVID 19 drug candidates. Following this path, the principal role of SPR biosensors relies on the recognition of drug targets that can interact with ACE2 receptors of infected cells or inhibit the S protein and MPro main protease of coronaviruses.
The identification of both natural compounds and authorized drugs requires the development of highly specialized biophysical methods. Although SPR configurations are perfectly suited for the screening of small molecules, the number of SPR applications in fragment-based drug discovery is still limited. SPR analysis can provide rapid and sensitive measurements with high selectivity and low protein consumption. Additionally, SPR biosensing can neutralize mass transport effects derived from protein immobilization thanks to the rapid dissociation and the low molecular weight of the analytes. Despite these remarkable advantages, several limitations may arise from the calculation of binding constants due to the low solubility of compounds, thereby requiring previous calibration in the solvent buffer. The multiplexing capacity is also lower in SPR devices in comparison with other biophysical methods such as NMR or X-ray crystallography. However, this limitation can be easily overcome by the low time of response of SPR biosensors. The structural information on binding sites can also be obtained by using competitive assay formats.
References
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154.
- Pattnaik, G.P.; Chakraborty, H. Entry Inhibitors: Efficient Means to Block Viral Infection. J. Membr. Biol. 2020, 253, 425–444.
- Sokolova, A.S.; Putilova, V.P.; Yarovaya, O.I.; Zybkina, A.V.; Mordvinova, E.D.; Zaykovskaya, A.V.; Shcherbakov, D.N.; Orshanskaya, I.R.; Sinegubova, E.O.; Esaulkova, I.L.; et al. Synthesis and Antiviral Activity of Camphene Derivatives against Different Types of Viruses. Molecules 2021, 26, 2235.
- Vigant, F.; Santos, N.C.; Lee, B. Broad-Spectrum Antivirals against Viral Fusion. Nat. Rev. Microbiol. 2015, 13, 426–437.
- Kumar, P.K.R. Systematic Screening of Viral Entry Inhibitors Using Surface Plasmon Resonance. Rev. Med. Virol. 2017, 27, e1952.
- Wang, Q.; Liu, Z. Recent Progress of Surface Plasmon Resonance in the Development of Coronavirus Disease-2019 Drug Candidates. Eur. J. Med. Chem. Rep. 2021, 1, 100003.
- Brouwer, P.J.M.; Caniels, T.G.; van der Straten, K.; Snitselaar, J.L.; Aldon, Y.; Bangaru, S.; Torres, J.L.; Okba, N.M.A.; Claireaux, M.; Kerster, G.; et al. Potent Neutralizing Antibodies from COVID-19 Patients Define Multiple Targets of Vulnerability. Science 2020, 369, 643–650.
- Shepherd, C.A.; Hopkins, A.L.; Navratilova, I. Fragment Screening by SPR and Advanced Application to GPCRs. Prog. Biophys. Mol. Biol. 2014, 116, 113–123.
- Congreve, M.; Rich, R.L.; Myszka, D.G.; Figaroa, F.; Siegal, G.; Marshall, F.H. Fragment Screening of Stabilized G-Protein-Coupled Receptors Using Biophysical Methods. Methods Enzym. 2011, 493, 115–136.
- Vanwetswinkel, S.; Heetebrij, R.J.; van Duynhoven, J.; Hollander, J.G.; Filippov, D.V.; Hajduk, P.J.; Siegal, G. TINS, Target Immobilized NMR Screening: An Efficient and Sensitive Method for Ligand Discovery. Chem. Biol. 2005, 12, 207–216.
- Blundell, T.L.; Jhoti, H.; Abell, C. High-Throughput Crystallography for Lead Discovery in Drug Design. Nat. Rev. Drug. Discov. 2002, 1, 45–54.
- Duong-Thi, M.-D.; Bergström, M.; Fex, T.; Isaksson, R.; Ohlson, S. High-Throughput Fragment Screening by Affinity LC-MS. J. Biomol. Screen. 2013, 18, 160–171.
- Ladbury, J.E.; Klebe, G.; Freire, E. Adding Calorimetric Data to Decision Making in Lead Discovery: A Hot Tip. Nat. Rev. Drug Discov. 2010, 9, 23–27.
- Kranz, J.K.; Schalk-Hihi, C. Protein Thermal Shifts to Identify Low Molecular Weight Fragments. Methods Enzymol. 2011, 493, 277–298.
- Lewis, L.M.; Engle, L.J.; Pierceall, W.E.; Hughes, D.E.; Shaw, K.J. Affinity Capillary Electrophoresis for the Screening of Novel Antimicrobial Targets. J. Biomol. Screen. 2004, 9, 303–308.
- Duong-Thi, M.-D.; Meiby, E.; Bergström, M.; Fex, T.; Isaksson, R.; Ohlson, S. Weak Affinity Chromatography as a New Approach for Fragment Screening in Drug Discovery. Anal. Biochem. 2011, 414, 138–146.
- Kaminski, T.; Gunnarsson, A.; Geschwindner, S. Harnessing the Versatility of Optical Biosensors for Target-Based Small-Molecule Drug Discovery. ACS Sens. 2017, 2, 10–15.
- Gauglitz, G. Critical Assessment of Relevant Methods in the Field of Biosensors with Direct Optical Detection Based on Fibers and Waveguides Using Plasmonic, Resonance, and Interference Effects. Anal. Bioanal. Chem. 2020, 412, 3317–3349.
- Olaru, A.; Bala, C.; Jaffrezic-Renault, N.; Aboul-Enein, H.Y. Surface Plasmon Resonance (SPR) Biosensors in Pharmaceutical Analysis. Crit. Rev. Anal. Chem. 2015, 45, 97–105.
- Shrivastav, A.M.; Cvelbar, U.; Abdulhalim, I. A Comprehensive Review on Plasmonic-Based Biosensors Used in Viral Diagnostics. Commun. Biol. 2021, 4, 70.
- Homola, J. Surface Plasmon Resonance Sensors for Detection of Chemical and Biological Species. Chem. Rev. 2008, 108, 462–493.
- Masson, J.-F. Surface Plasmon Resonance Clinical Biosensors for Medical Diagnostics. ACS Sens. 2017, 2, 16–30.
- Lin, S.; Shih-Yuan Lee, A.; Lin, C.-C.; Lee, C.-K. Determination of Binding Constant and Stoichiometry for Antibody-Antigen Interaction with Surface Plasmon Resonance. Curr. Proteom. 2006, 3, 271–282.
- Chavanieu, A.; Pugnière, M. Developments in SPR Fragment Screening. Expert Opin. Drug Discov. 2016, 11, 489–499.
- Cao, Y.; Cao, Y.; Shi, Y.; Cai, Y.; Chen, L.; Wang, D.; Liu, Y.; Chen, X.; Zhu, Z.; Hong, Z.; et al. Surface Plasmon Resonance Biosensor Combined with Lentiviral Particle Stabilization Strategy for Rapid and Specific Screening of P-Glycoprotein Ligands. Anal. Bioanal. Chem. 2021, 413, 2021–2031.
- Giannetti, A.M. From Experimental Design to Validated Hits a Comprehensive Walk-through of Fragment Lead Identification Using Surface Plasmon Resonance. Methods Enzymol. 2011, 493, 169–218.
- Mazzon, M.; Marsh, M. Targeting Viral Entry as a Strategy for Broad-Spectrum Antivirals. F1000Research 2019, 8. F1000 Faculty Rev-1628.
- Hajduk, P.J.; Greer, J. A Decade of Fragment-Based Drug Design: Strategic Advances and Lessons Learned. Nat. Rev. Drug Discov. 2007, 6, 211–219.
- Pollack, S.J.; Beyer, K.S.; Lock, C.; Müller, I.; Sheppard, D.; Lipkin, M.; Hardick, D.; Blurton, P.; Leonard, P.M.; Hubbard, P.A.; et al. A Comparative Study of Fragment Screening Methods on the P38α Kinase: New Methods, New Insights. J. Comput. Aided Mol. Des. 2011, 25, 677–687.
- Boettcher, A.; Ruedisser, S.; Erbel, P.; Vinzenz, D.; Schiering, N.; Hassiepen, U.; Rigollier, P.; Mayr, L.M.; Woelcke, J. Fragment-Based Screening by Biochemical Assays: Systematic Feasibility Studies with Trypsin and MMP12. J. Biomol. Screen. 2010, 15, 1029–1041.
- Rouhana, J.; Hoh, F.; Estaran, S.; Henriquet, C.; Boublik, Y.; Kerkour, A.; Trouillard, R.; Martinez, J.; Pugnière, M.; Padilla, A.; et al. Fragment-Based Identification of a Locus in the Sec7 Domain of Arno for the Design of Protein-Protein Interaction Inhibitors. J. Med. Chem. 2013, 56, 8497–8511.
- Zhu, Z.-L.; Qiu, X.-D.; Wu, S.; Liu, Y.-T.; Zhao, T.; Sun, Z.-H.; Li, Z.-R.; Shan, G.-Z. Blocking Effect of Demethylzeylasteral on the Interaction between Human ACE2 Protein and SARS-CoV-2 RBD Protein Discovered Using SPR Technology. Molecules 2020, 26, 57.
- Day, C.J.; Bailly, B.; Guillon, P.; Dirr, L.; Jen, F.E.-C.; Spillings, B.L.; Mak, J.; von Itzstein, M.; Haselhorst, T.; Jennings, M.P. Multidisciplinary Approaches Identify Compounds That Bind to Human ACE2 or SARS-CoV-2 Spike Protein as Candidates to Block SARS-CoV-2–ACE2 Receptor Interactions. mBio 2021, 12, e03681-20.
- Xu, H.; Liu, B.; Xiao, Z.; Zhou, M.; Ge, L.; Jia, F.; Liu, Y.; Jin, H.; Zhu, X.; Gao, J.; et al. Computational and Experimental Studies Reveal That Thymoquinone Blocks the Entry of Coronaviruses Into In Vitro Cells. Infect Dis. Ther. 2021, 10, 483–494.
- Yu, S.; Zhu, Y.; Xu, J.; Yao, G.; Zhang, P.; Wang, M.; Zhao, Y.; Lin, G.; Chen, H.; Chen, L.; et al. Glycyrrhizic Acid Exerts Inhibitory Activity against the Spike Protein of SARS-CoV-2. Phytomedicine 2021, 85, 153364.
- Mei, J.; Zhou, Y.; Yang, X.; Zhang, F.; Liu, X.; Yu, B. Active Components in Ephedra Sinica Stapf Disrupt the Interaction between ACE2 and SARS-CoV-2 RBD: Potent COVID-19 Therapeutic Agents. J. Ethnopharmacol. 2021, 278, 114303.
- Gao, J.; Ding, Y.; Wang, Y.; Liang, P.; Zhang, L.; Liu, R. Oroxylin A Is a Severe Acute Respiratory Syndrome Coronavirus 2-spiked Pseudotyped Virus Blocker Obtained from Radix Scutellariae Using Angiotensin-converting Enzyme II /Cell Membrane Chromatography. Phytother. Res. 2021, 35, 3194–3204.
- Binette, V.; Côté, S.; Haddad, M.; Nguyen, P.T.; Bélanger, S.; Bourgault, S.; Ramassamy, C.; Gaudreault, R.; Mousseau, N. Corilagin and 1,3,6-Tri- O -Galloy-β- D -Glucose: Potential Inhibitors of SARS-CoV-2 Variants. Phys. Chem. Chem. Phys. 2021, 23, 14873–14888.
- Singh, S.K.; Singh, S.; Singh, R. Targeting Novel Coronavirus SARS-CoV-2 Spike Protein with Phytoconstituents of Momordica Charantia. J. Ovarian Res. 2021, 14, 126.
- Ge, S.; Wang, X.; Hou, Y.; Lv, Y.; Wang, C.; He, H. Repositioning of Histamine H1 Receptor Antagonist: Doxepin Inhibits Viropexis of SARS-CoV-2 Spike Pseudovirus by Blocking ACE2. Eur. J. Pharmacol. 2021, 896, 173897.
- Ge, S.; Lu, J.; Hou, Y.; Lv, Y.; Wang, C.; He, H. Azelastine Inhibits Viropexis of SARS-CoV-2 Spike Pseudovirus by Binding to SARS-CoV-2 Entry Receptor ACE2. Virology 2021, 560, 110–115.
- Hou, Y.; Ge, S.; Li, X.; Wang, C.; He, H.; He, L. Testing of the Inhibitory Effects of Loratadine and Desloratadine on SARS-CoV-2 Spike Pseudotyped Virus Viropexis. Chem.-Biol. Interact. 2021, 338, 109420.
- Wang, N.; Han, S.; Liu, R.; Meng, L.; He, H.; Zhang, Y.; Wang, C.; Lv, Y.; Wang, J.; Li, X.; et al. Chloroquine and Hydroxychloroquine as ACE2 Blockers to Inhibit Viropexis of 2019-NCoV Spike Pseudotyped Virus. Phytomedicine 2020, 79, 153333.
- Lu, J.; Hou, Y.; Ge, S.; Wang, X.; Wang, J.; Hu, T.; Lv, Y.; He, H.; Wang, C. Screened Antipsychotic Drugs Inhibit SARS-CoV-2 Binding with ACE2 in Vitro. Life Sci. 2021, 266, 118889.
- Li, H.; Xu, F.; Liu, C.; Cai, A.; Dain, J.A.; Li, D.; Seeram, N.P.; Cho, B.P.; Ma, H. Inhibitory Effects and Surface Plasmon Resonance-Based Binding Affinities of Dietary Hydrolyzable Tannins and Their Gut Microbial Metabolites on SARS-CoV-2 Main Protease. J. Agric. Food Chem. 2021, 69, 12197–12208.

