 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vladimir Muronetz | + 1716 word(s) | 1716 | 2021-11-21 08:17:05 | | | |
| 2 | Vicky Zhou | Meta information modification | 1716 | 2021-12-03 03:10:48 | | |
Video Upload Options
One of the main targets of NO in cells is glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which is due to the presence of a highly reactive cysteine residue in the active center of this protein, as well as to the high content of this protein in all cells. The main products of GAPDH modification with nitric oxide are S-nitrosylated GAPDH and S-sulfenated GAPDH (GAPDH-SNO and GAPDH-SOH, respectively). Modification of GAPDH with nitric oxide is of particular interest due to the fact that it causes the accumulation of GAPDH in the nucleus and induction of apoptosis. According to the most popular hypothesis, a wide range of apoptotic stimuli augment NO production in the cells, GAPDH is nitrosylated by NO, which leads to inactivation of the protein and to conformational changes in its molecule. These alterations facilitate the binding of GAPDH with the E3-ubiquitin-ligase Siah1. Siah1, which possesses a nuclear localization signal (NLS), translocates GAPDH to the nucleus and stimulates a cascade of apoptotic reactions. However, the molecular mechanisms of this signaling pathway have not yet been studied in detail. There are no direct data on the selective interaction of the S-nitrosylated GAPDH with Siah1. It cannot be excluded that any modifications of the catalytic cysteine residue, which are accompanied by a weakening of interactions between the enzyme and NAD+, lead to a change in the conformation of GAPDH, and stimulate its binding to some partner proteins—in particular, Siah1.
1. Introduction
Sulfhydryl groups of cysteine residues can undergo S-nitrosylation to form the corresponding nitrosothiols (Protein-S-NO). S-nitrosylation is considered to be an important post-translational modification of proteins involved in physiological regulation based on redox potential [1][2][3][4]. One of the main targets of NO in cells is glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which is due to the presence of a highly reactive cysteine residue in the active center of this protein, as well as to the high content of this protein in all cells (5–15% of the total amount of soluble proteins of the cytoplasm) [5][6]. Post-translational modifications of GAPDH are of special interest, since they can affect not only the catalytic activity of the enzyme, but also its numerous moonlighting functions including the induction of apoptosis [7]. Modifications of GAPDH can influence its interactions with other proteins and RNA, which affect the development of human disease, tumorigenesis, diabetes, and age-related neurodegenerative disorders [8].
Studies of the effect of nitric oxide on GAPDH have been taking place for almost half a century, but the mechanisms of this modification, and its consequences for the vital activity of the cell, are still far from being fully understood. In 1973, it was first discovered that organic nitrates inhibit the activity of GAPDH and monoamine oxidase—probably due to the modification of sulfhydryl groups important for catalysis [9]. Later, it was shown that the treatment of GAPDH with trinitroglycerin resulted in the disappearance of the main dehydrogenase activity with simultaneous development of the acylphosphatase activity [10]. The authors suggested that this modification was due to the oxidation of the catalytic cysteine residue to cysteine sulfenic acid. Thus, this study did not suggest the S-nitrosylation of GAPDH, and did not consider a role of such a modification in the regulation of the enzyme function.
In 1992, three groups of researchers suggested that NO stimulates ADP-ribosylation of GAPDH [11][12][13]. Later, it was proven that the entire NAD+ molecule binds to the enzyme. This process is indeed stimulated by nitric oxide, which nitrosylates the cysteine residue in the active center of GAPDH, leading to the inactivation of the enzyme and the covalent binding of NAD+. However, the inactivation of GAPDH mainly occurs due to S-nitrosylation of the catalytic cysteine residue, since the covalent inclusion of NAD+ does not exceed 0.02 mol NAD+ per mol GAPDH monomer [14]. Since then, it was believed that the product of the modification of GAPDH by NO is GAPDH nitrosylated at the catalytic cysteine residue (GAPDH-SNO). Recently, it has been shown that the main products of GAPDH modification with nitric oxide are S-nitrosylated GAPDH and S-sulfenated GAPDH (GAPDH-SNO and GAPDH-SOH, respectively) [15]. Consequently, S-nitrosylation of GAPDH results in the oxidation of the catalytic cysteine, yielding the relatively stable cysteine sulfenic acid, which cannot exclude its further oxidation:
 Scheme 1. Relationship between S-nitrosylation and oxidation of GAPDH.
Scheme 1. Relationship between S-nitrosylation and oxidation of GAPDH.
2. Development of Ideas about the Induction of Apoptosis with the Participation of GAPDH.
Since the mid-1990s, information has begun to accumulate about the participation of GAPDH in the induction of apoptosis. It was shown that in various pathologies, this cytoplasmic enzyme accumulated in the nucleus, which correlated with the induction of apoptosis [16][17]. This accumulation of GAPDH in the nucleus could be caused by exposure to nitric oxide, reactive oxygen species, and other compounds [17][18][19]. Special attention to the nuclear translocation of GAPDH was prompted by the relationship of this phenomenon to the development of neurodegenerative disorders—primarily Parkinson's disease. However, ideas about the mechanism of the translocation of GAPDH into the nucleus were contradictory. It is well known that GAPDH is a tetrameric protein of 144 kDa localized in the cytoplasm. The large size of the protein excludes its translocation into the nucleus in the native state. The transport of the GAPDH tetramer to the nucleus via carrier proteins is impossible, since the enzyme lacks the nuclear localization signal (NLS). Therefore, the mechanism of GAPDH translocation to the nucleus requires a stage of the dissociation of the S-nitrosylated, oxidized, or otherwise modified GAPDH tetramer into subunits. In contrast to the tetrameric molecule, subunits can penetrate into the cell nucleus via passive transport [16]. The assumption of the dissociation of a tetrameric molecule into subunits upon modification of the catalytic cysteine residue is based on the fact that NAD+ is tightly bound in the active center of GAPDH and exerts a pronounced stabilizing effect on the protein. Oxidation and other modifications of the catalytic cysteine residue decrease the affinity of NAD+ for the protein. Consequently, the release of NAD+ from the active center can induce the dissociation of the tetramer into subunits, and their subsequent movement into the nucleus (Figure 1). In addition, the oxidized forms of GAPDH have an increased affinity for nucleic acids, the interaction with which in the nucleus is considered to be one of the mechanisms of apoptosis [20].
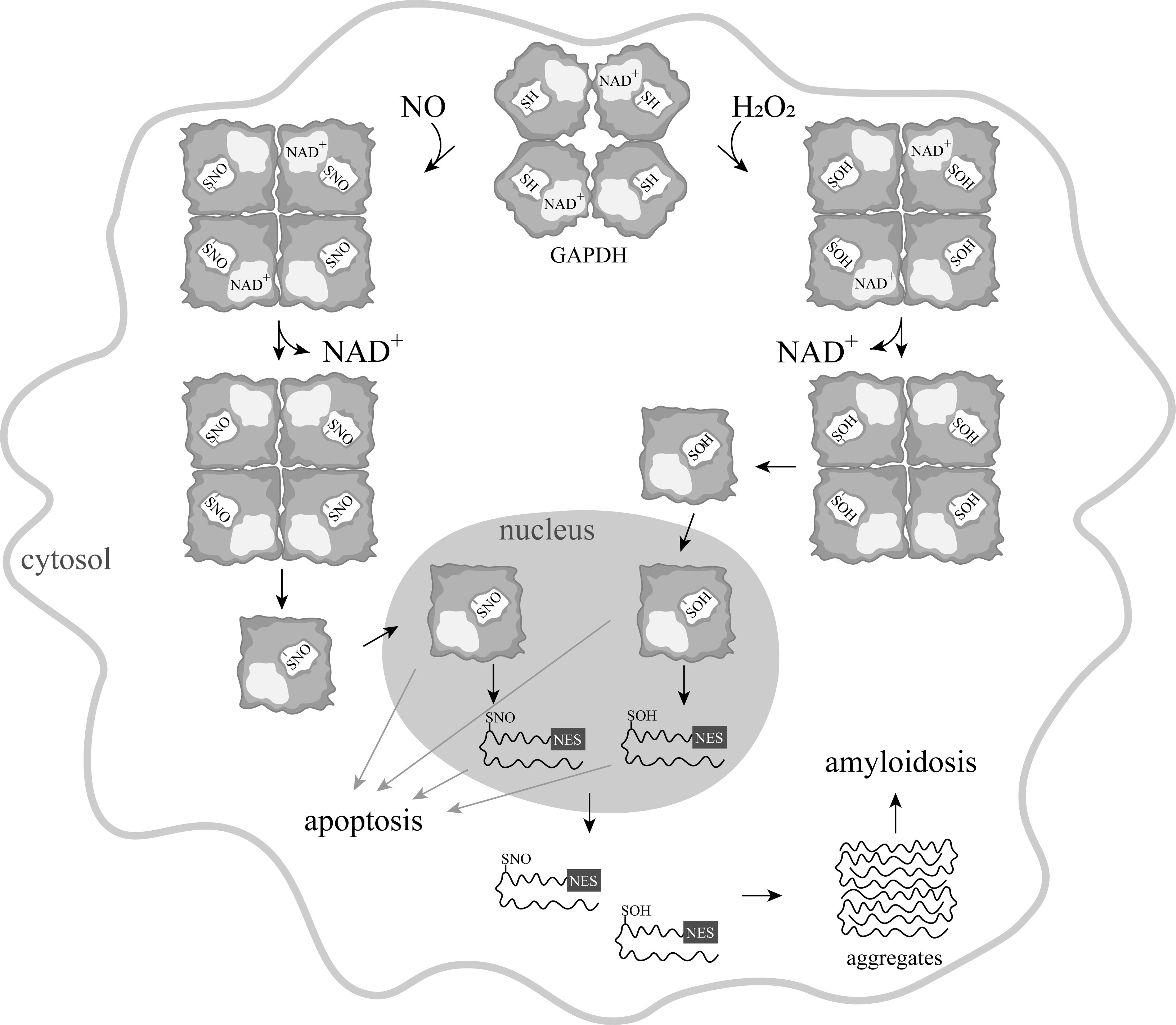
Figure 1. A scheme of apoptosis induced by various modifications of GAPDH.
In 2006, a new concept was proposed according to which the main link in the NO-induced apoptosis is S-nitrosylation of GAPDH. According to this idea, a wide range of apoptotic stimuli augment NO production via the induction of inducible nitric oxide synthase (iNOS), or by the activation of neuronal nitric oxide synthase (nNOS). GAPDH is nitrosylated by NO, which leads to inactivation of the protein and to conformational changes in its molecule. These alterations facilitate the binding of GAPDH with the E3-ubiquitin-ligase Siah1. Siah1, which possesses a nuclear localization signal (NLS), translocates GAPDH to the nucleus and stimulates a cascade of apoptotic reactions (Figure 2) [21].
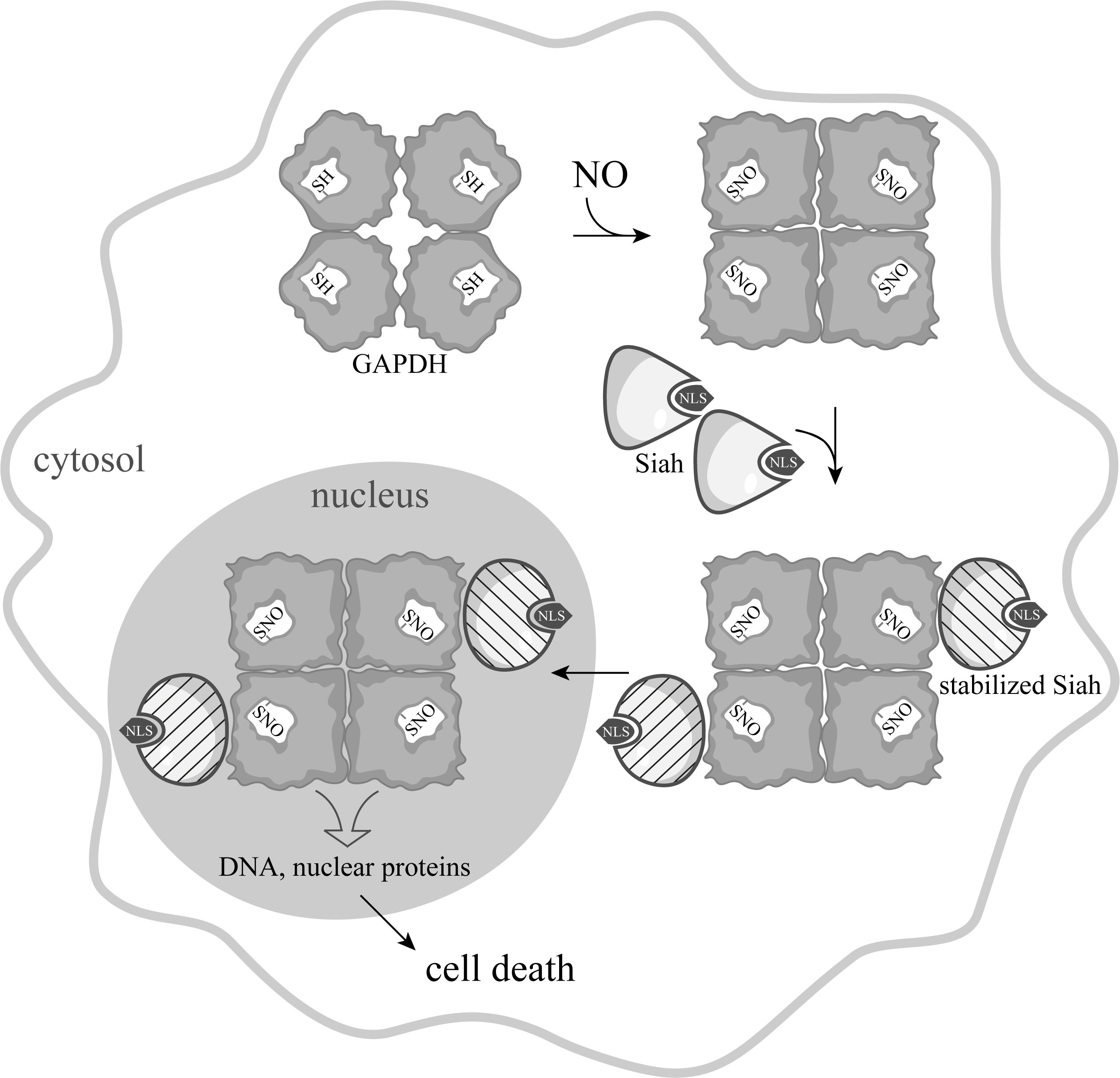
Figure 2. Scheme of NO-induced apoptosis mediated by GAPDH proposed by A. Sawa [22], with the depiction of the tetrameric GAPDH structure.
The mechanism by which nuclear GAPDH triggers apoptosis remains unclear. Initially it was assumed that the interaction with GAPDH results in the stabilization of Siah1, which enables rapid degradation of nuclear proteins, leading to cell death [22][23][24]. According to a later version of this mechanism, nuclear GAPDH is acetylated at Lys 160 by the acetyltransferase p300/CBP (CREB-binding protein) via direct protein interaction, which, in turn, stimulates the acetylation and catalytic activity of p300/CBP, activation of the down-stream targets of p300/CBP (such as p53), and cause cell death [25]. One more mechanism of signal transduction mediated by S-nitrosylated GAPDH was proposed, according to which the nitric oxide group is transferred from S-nitrosylated GAPDH to nuclear proteins, including the deacetylating enzyme sirtuin-1 (SIRT1), histone deacetylase-2 (HDAC2), and DNA-activated protein kinase (DNA-PK) [26]. Besides, using rat stroke model, it was shown that nuclear GAPDH binds with poly(ADP-ribose) polymerase-1 (PARP-1), and this complex promotes PARP-1 overactivation, leading to brain damage and neurological deficits [27].
3. Induction of Apoptosis by Oxidation and Other Modifications of the Catalytic Cysteines of GAPDH
Some facts indicate that S-nitrosylation of GAPDH is not the only mechanism for the induction of apoptosis with the participation of GAPDH. This assumption is confirmed by the observation on the direct induction of apoptosis by hydrogen peroxide [19]. It cannot be excluded that GAPDH with oxidized sulfhydryl groups moves into the nucleus and causes a cascade of reactions, leading to apoptosis. It was assumed that the oxidation of cysteine residues leads to a weakening of NAD+ binding in the active center of the enzyme, which results in its dissociation from the active center, decreasing the stability of the tetrameric molecule. Monomeric subunits of GAPDH of 36 kDa can penetrate into the nucleus due to passive transport. In the nucleus, further unfolding of subunits and exposure of the nuclear export signal (NES) take place. Such unfolded GAPDH subunits are transported into the cytoplasm or released from the cells broken during apoptosis, aggregate, and exert a toxic effect on cells unaffected by apoptosis (Fig. 1).
4. Questions to be Resolved
The induction of apoptosis by NO through a signaling pathway, the key element of which is GAPDH, has been confirmed in many studies. However, the molecular mechanisms of this signaling pathway have not yet been studied in detail. Some aspects of the described model of apoptosis remain unclear.
1) There are no direct data on the selective interaction of the S-nitrosylated GAPDH (GAPDH-SNO) with partner proteins. Most likely, any modifications of the catalytic cysteine residue, which are accompanied by a weakening of interactions between the enzyme and NAD+, lead to a change in the conformation of GAPDH, and stimulate its binding to some partner proteins—in particular, Siah1.
2) The mechanism of the influence of S-nitrosylation of GAPDH on its ability to interact with Siah1 is unclear. There is no precise information on the effect of S-nitrosylation on the enzyme structure, or on the necessity of S-nitrosylation of GAPDH for the implementation of the cascade of processes that induce apoptosis.
3) There is no direct evidence on the interaction of the GAPDH molecule with Siah1, nor on the enhancement of this interaction after S-nitrosylation of GAPDH. It remains unclear whether it is a tetrameric GAPDH molecule that is involved in this signaling pathway, or whether Siah1 interacts with a separate polypeptide chain of GAPDH.
References
- Hess Douglas, Matsumoto Akio, Kim Sung-Oog, Marshall Harvey, Stamler Jonathan; Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol . 2005, 6(2), 150-66, DOI: 10.1038/nrm1569.
- Foster, M.W.; Hess, D.T.; Stamler, J.S; Protein S-nitrosylation in health and disease: a current perspective. Trends Mol. Med. 2009, 15, 391–404, https://doi.org/10.1016/j.molmed.2009.06.007.
- Tossounian, M.A.; Zhang, B.; Gout, I.; The writers, readers, and erasers in redox regulation of GAPDH. Antioxidants 2020, 9, 1288, https://doi.org/10.3390/antiox9121288.
- Muronetz, V.I.; Melnikova, A.K.; Saso, L.; Schmalhausen, E.V.; Influence of Oxidative Stress on Catalytic and Non-glycolytic Functions of Glyceraldehyde-3-phosphate Dehydrogenase. Curr. Med. Chem. 2020, 27, 2040–2058, DOI: 10.2174/0929867325666180530101057.
- Seidler, N.W. GAPDH: Biological Properties and Diversity; Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2013; pp. Volume 985.
- Harris, J.I.; Waters, M.. Glyceraldehyde 3-Phosphate Dehydrogenase. In The Enzymes, 3rd ed.; Oxidation-Reduction Part C; Boyer, P.D., Eds.; Academic Press: London, UK, 1976; pp. Volume XIII, pp. 1–49.
- Sirover, M.A.; On the functional diversity of glyceraldehyde-3-phosphate dehydrogenase: Biochemical mechanisms and regulatory control. Biochim. Biophys. Acta. 2011, 1810, 741–751, DOI: 10.1016/j.bbagen.2011.05.010.
- Sirover, M.A.; Moonlighting glyceraldehyde-3-phosphate dehydrogenase: Posttranslational modification, protein and nucleic acid interactions in normal cells and in human pathology. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 354–371, https://doi.org/10.1080/10409238.2020.1787325.
- Jakschik, S.B.; Needleman, P.; Sulfhydryl reactivity of organic nitrates: Biochemical basis for inhibition of glyceraldehyde-P dehydrogenase and monoamine oxidase. Biochem. Biophys. Res. Commun. 1973, 53, 539–544, https://doi.org/10.1016/0006-291X(73)90695-5.
- You, K.-S.; Benitez, L.V.; McConachie, W.A.; Allison, W.S.; The conversion of glyceraldehyde-3-phosphate dehyrogenase to an acylphosphatase by trinitroglycerin and inactivation of this activity by azide and ascorbate.. Biochim. Biophys. Acta-Enzymol. 1975, 384, 317–330, DOI: 10.1016/0005-2744(75)90033-9.
- Kots, A.Y.; Skurat, A.V.; Sergienko, E.A.; Bulargina, T.V.; Severin, E.S.; Nitroprusside stimulates the cysteine-specific mono(ADP-ribosylation) of glyceraldehyde-3-phosphate dehydrogenase from human erythrocytes.. FEBS Lett. 1992, 300, 9–12, https://doi.org/10.1016/0014-5793(92)80153-8.
- Dimmeler, S.; Lottspeich, F.; Brüne, B.; Nitric oxide causes ADP-ribosylation and inhibition of glyceraldehyde-3-phosphate dehydrogenase.. J. Biol. Chem. 1992, 267, 16771–16774, PMID: 1512218.
- Zhang, J.; Snyder, S.H.; Nitric oxide stimulates auto-ADP-ribosylation of glyceraldehyde-3-phosphate dehydrogenase.. Proc. Natl. Acad. Sci. USA 1992, 89, 9382–9385, DOI: 10.1073/pnas.89.20.9382.
- McDonald, L.J.; Moss, J.; Stimulation by nitric oxide of an NAD linkage to glyceraldehyde-3-phosphate dehydrogenase.. Proc. Natl. Acad. Sci. USA 1993, 90, 6238–6241, https://doi.org/10.1073/pnas.90.13.6238.
- Schmalhausen, E.V.; Medvedeva, M.V.; Serebryakova, M.V.; Chagovets, V.V.; Muronetz, V.I.; Products of S-nitrosylation of glyceraldehyde-3-phosphate dehydrogenase: Relation between S-nitrosylation and oxidation.. Biochim. Biophys. Acta-Gen. Subj. 2021, 1866, 130032, DOI: 10.1016/j.bbagen.2021.130032.
- Carlile, G.W.; Chalmers-Redman, R.M.; Tatton, N.A.; Borden, K.E.; Tatton, W.G.; Reduced apoptosis after nerve growth factor and serum withdrawal: Conversion of tetrameric glyceraldehyde-3-phosphate dehydrogenase to a dimer.. Mol. Pharmacol. 2001, 57, 2–12, PMID: 10617673.
- Brown, V.M.; Krynetski, E.Y.; Krynetskaia, N.F.; Grieger, D.; Mukatira, S.T.; Murti, K.G.; Slaughter, C.A.; Park, H.-W.; Evans, W.E.; A Novel CRM1-mediated Nuclear Export Signal Governs Nuclear Accumulation of Glyceraldehyde-3-phosphate Dehydrogenase following Genotoxic Stress.. J. Biol. Chem. 2004, 279, 5984–5992, DOI: 10.1074/jbc.M307071200.
- Dastoor, Z.; Dreyer, J.L.; Potential role of nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase in apoptosis and oxidative stress.. J. Cell Sci. 2001, 114, 1643–1653, PMID: 11309196.
- Arutyunova, E.I.; Domnina, L.V.; Chudinova, A.A.; Makshakova, O.N.; Arutyunov, D.Y.; Muronetz, V.I.; Localization of non-native D-glyceraldehyde-3-phosphate dehydrogenase in growing and apoptotic HeLa cells.. Biochemistry 2013, 78, 91–95, DOI: 10.1134/S0006297913010112.
- Arutyunova, E.I.; Danshina, P.V.; Domnina, L.V.; Pleten, A.P.; Muronetz, V.I.; Oxidation of glyceraldehyde-3-phosphate dehydrogenase enhances its binding to nucleic acids.. Biochem. Biophys. Res. Commun. 2003, 307, 547-552, DOI: 10.1016/s0006-291x(03)01222-1.
- Hara, M.R.; Snyder, S.H.; Nitric Oxide–GAPDH–Siah: A Novel Cell Death Cascade.. Cell. Mol. Neurobiol. 2006, 26, 527–538, DOI: 10.1007/s10571-006-9011-6.
- Hara, M.R.; Cascio, M.B.; Sawa, A.; GAPDH as a sensor of NO stress.. Biochim. Biophys. Acta-Mol. Basis Dis. 2006, 1762, 502–509, DOI: 10.1016/j.bbadis.2006.01.012.
- Hara, M.R.; Snyder, S.H.; Cell signaling and neuronal death.. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 117–141, DOI: 10.1146/annurev.pharmtox.47.120505.105311.
- Hara, M.R.; Thomas, B.; Cascio, M.B.; Bae, B.-I.; Hester, L.D.; Dawson, V.L.; Dawson, T.M.; Sawa, A.; Snyder, S.H.; Neuroprotection by pharmacologic blockade of the GAPDH death cascade.. Proc. Natl. Acad. Sci. USA 2006, 103, 3887–3889, DOI: 10.1073/pnas.0511321103.
- Sen, N.; Hara, M.R.; Kornberg, M.D.; Cascio, M.B.; Bae, B.-I.; Shahani, N.; Thomas, B.; Dawson, T.M.; Dawson, V.L.; Snyder, S.H.; et al.Sawa A. Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis.. Nat. Cell Biol. 2008, 10, 866–873, DOI: 10.1038/ncb1747.
- Kornberg, M.D.; Sen, N.; Hara, M.R.; Juluri, K.R.; Nguyen, J.V.K.; Snowman, A.M.; Law, L.; Hester, L.D.; Snyder, S.H.; GAPDH mediates nitrosylation of nuclear proteins.. Nat. Cell Biol. 2010, 12, 1094–1100, DOI: 10.1038/ncb2114.
- Hidemitsu Nakajima; Takeya Kubo; Hideshi Ihara; Takatoshi Hikida; Teruko Danjo; Masatoshi Nakatsuji; Neelam Shahani; Masanori Itakura; Yoko Ono; Yasu-Taka Azuma; et al.Takashi InuiAtsushi KamiyaAkira SawaTadayoshi Takeuchi Nuclear-translocated Glyceraldehyde-3-phosphate Dehydrogenase Promotes Poly(ADP-ribose) Polymerase-1 Activation during Oxidative/Nitrosative Stress in Stroke.. J. Biol. Chem. 2015, 290, 14493–14503, doi: 10.1074/jbc.M114.635607.


