Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lawrence Boise | + 2100 word(s) | 2100 | 2021-10-22 08:51:38 | | | |
| 2 | Jessie Wu | Meta information modification | 2100 | 2021-12-03 09:17:55 | | | | |
| 3 | Jessie Wu | + 4 word(s) | 2104 | 2021-12-03 09:21:12 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Boise, L. Chimeric Antigen Receptor T cells in Multiple Myeloma. Encyclopedia. Available online: https://encyclopedia.pub/entry/16565 (accessed on 25 June 2026).
Boise L. Chimeric Antigen Receptor T cells in Multiple Myeloma. Encyclopedia. Available at: https://encyclopedia.pub/entry/16565. Accessed June 25, 2026.
Boise, Lawrence. "Chimeric Antigen Receptor T cells in Multiple Myeloma" Encyclopedia, https://encyclopedia.pub/entry/16565 (accessed June 25, 2026).
Boise, L. (2021, November 30). Chimeric Antigen Receptor T cells in Multiple Myeloma. In Encyclopedia. https://encyclopedia.pub/entry/16565
Boise, Lawrence. "Chimeric Antigen Receptor T cells in Multiple Myeloma." Encyclopedia. Web. 30 November, 2021.
Copy Citation
Multiple myeloma is a disease of malignant plasma cells and the second most common hematological cancer. This entry describes the history and use of CAR-T cells for the treatment of this disease as well as comment on future approaches.
multiple myeloma
immunotherapy
CAR-T
1. CAR-T Cells
Chimeric Antigen Receptor (CAR) T cells have shown unprecedented response rates in hematological cancers. Since 2017, several CAR-T therapies have been approved by the FDA for pediatric leukemia and adult lymphomas and, just this year, the first CAR-T therapy against multiple myeloma was approved. In the current paradigm, the patient’s own T cells are virally modified to introduce a chimeric receptor directed towards surface molecules of the target tumor cells. This use of an autologous therapy results in reduced concerns regarding rejection of the therapy and a greater potential for prolonged protection. Recent efforts are also focusing on allogenic CAR-Ts and gene-edited CAR-Ts.
2. Mechanism of Action
The engineered introduction of the CAR molecules into the T cells permits the targeting of tumor cells by pairing non-MHC-dependent molecular recognition with native T cell signaling. This allows for the same cascade to be triggered as when the T cell receptor (TCR) detects its cognate antigen (Figure 2) [1]. This mechanism results in effective targeted killing through granzyme and perforin secretion, similar to the native way in which T cells kill target cells [2]. Given that the targeting is based on either antibody fragments or catalogued libraries of ligand-binding motifs, there is a wide array of potential targets which can be developed into CAR constructs to treat various cancers [1]. Additionally, CAR-T cells have demonstrated killing through the use of the Fas/FasL pathway wherein FasL (CD95L) on the CAR-T cell binds with its receptor Fas (CD95) and induces activation of caspase 8, which forms a death-inducing signaling complex (DISC) in the targeted cell [2]. There data suggest that this mechanism is primarily used following antigen-positive activation of the CAR-T cells, which aids in the removal of antigen negative cancer. However, the threat of off-target killing increases as this pathway is more exploited by the therapeutic cells [3].
3. History
The initial case reports and successful clinical trials using CAR-T cells for the treatment of cancer consisted of targeting CD19 in cases of relapsed and refractory B cell malignancies [4]. These trials showed incredibly high rates of cancer remission when patients were given CAR-T therapies [5]. Occasionally, the delivered cell product was undetectable within a few months, which tended to result in disease relapse, given the loss of protection by the therapeutic cells. However, the authors of the study validated that the patient’s cancer cells were still sensitive to the same CAR-T cell-killing, meaning that reinfusion of the product or a more persistent cell product would provide protection and induce remission again [5]. Following these initial clinical trials, two anti-CD19 CAR-T cell products gained FDA approval in 2017: tisagenlecleucel from Novartis Pharmaceuticals following their B2202 trial and axicabtagene ciloleucel from Kite Pharma following their ZUMA-1 trial. These approvals further galvanized researchers and companies to operate their own trials using CAR-T to treat other forms of cancer.
4. CAR-T in Multiple Myeloma
As discussed above, multiple myeloma is one of several cancers for which a CAR-T product is now FDA-approved, and many other clinical trials are underway. The primary target that has been explored in the context of myeloma is the B-cell, maturation antigen (BCMA), given that it has minimal off-target expression (Figure 1) [6]. Additionally, BCMA is essential for plasma cell proliferation and survival. Therefore, it is unlikely to undergo sufficient mutation to allow the cancer cells to escape from the therapy [7]. However, there are also trials exploring the use of other targets, such as NY-ESO-1, CD-19, and GPRC5D [8][9][10]. NY-ESO-1 is a cancer/testis antigen that is highly specific to the cancer cells and has shown to be a useful CAR-T target, but it does require genetic sequencing of the multiple myeloma in order to validate that it is being expressed in the patient. Conversely, CD-19 is very rarely expressed in plasma cells, but CAR-T trials have validated that it seems to be an effective target in the context of multiple myeloma [9]. GPRC5D is also not canonically expressed in plasma cells but has been detected in multiple myeloma patients independent of BCMA expression, meaning that it could serve as a useful secondary target [10].
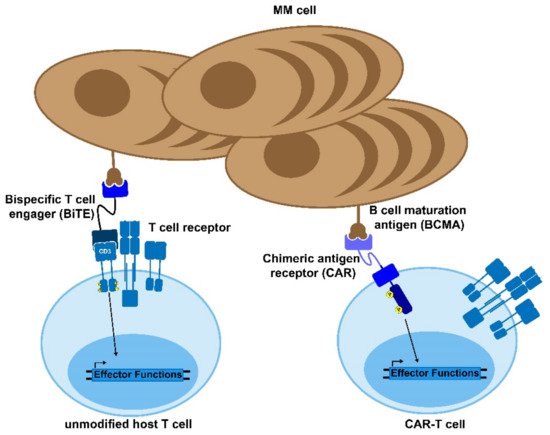
Figure 1. Bi-specific antibodies and CAR-T cells. Bispecific T cell engagers (BiTEs) and chimeric antigen receptor (CAR) T cells are novel therapies to treat MM and other diseases. The BiTE uses two antibody domains to crosslink the target cell to an immune effector cell in order to activate the host immune response against the tumor. Similarly, CAR-T cells are genetically modified to express a chimeric, MM-recognizing antigen that can bypass the normal T cell activation cascade to enable the CAR-T cell to respond to and kill target antigen-expressing cells.
In March of 2021, the FDA formally approved the first CAR-T therapy for relapsed/refractory multiple myeloma in idecabtagene vicleucel from Bristol Myers Squibb and bluebird bio, following a trial showing an overall response rate of ~70% in the phase 2 trial. The approval of idecabtagene vicleucel and its CD-19 predecessors tisagenlecleucel and axicabtagene ciloleucel further validate that CAR-T cell products will likely continue to gain approval for various cancer types.
5. Current Drawbacks to CAR-T
While the short-term response rates of CAR-T therapies tend to be encouraging, there is still a significant risk of post-treatment relapse. The primary concern is that CAR-T treatments for multiple myeloma tend to have problems with persistence, where the T cells do not engraft and therefore do not provide prolonged protection. This is emphasized by the fact that in vivo expansion is typically the most consistent factor, correlating with patient responses across clinical trials [11]. The working hypothesis for this issue is that the manufactured T cells display a proportion of memory subphenotypes that is too small to persist and engraft [12]. Another likely cause of post-therapy remission is exhaustion of the CAR-T cells due to overactivation during expansion or anergy following delivery [13]. This is particularly due to the immunosuppressive microenvironment seen in the case of multiple myeloma but not in other diseases for which CAR-T therapy is approved. Antigen loss, where the relapsed disease does not express the original CAR target, is another source of concern as well [14][15]. This is more commonly seen in the case of CD-19-targeting therapies, but recent publications have shown biallelic deletion of BCMA, which have caused therapy to fail upon administration of a second CAR-T infusion [16]. This outcome currently represents a minority of cases in multiple myeloma, as the majority of patients relapse with BCMA+ disease, but is still being considered as more data become available from clinical trials [11]. Finally, cancer dysregulation of death receptor signaling, such as FADD, BID, and TRAIL2, has been shown to play a role in CAR-T cell off-target killing. However, its effects in the context of multiple myeloma are still under investigation [3].
There are two key clinical toxicities that have been noted with the delivery of CAR-Ts: CRS and neurotoxicity [17]. While the clinical symptoms of the toxicities vary and can range from mild to life-threatening, the prevailing hypothesis is that both are caused by the same phenomenon. The rapid expansion and activation of the delivered CAR-T cells results in a sudden increase in immunostimulatory cytokines, such as TNF, IFN-γ, and GM-CSF, which activate innate immune cells and trigger a positive feedback loop of these same cytokines, called a cytokine storm [17]. The likelihood and grade of these toxicities is correlated with patient tumor burden, requiring close monitoring of these high-risk patients following treatment administration [4][17]. The clinical management of these toxicities is commonly carried out through the use of immunosuppressive agents such as corticosteroids, cytokine receptor antagonists (anakinra), and antibodies targeting either the cytokines themselves (lenzilumab) or their receptor (tocilizumab) [17].
The bone marrow niche where multiple myeloma proliferates and differentiates is a complex interaction of cells, cytokines, and a noncellular compartment [18]. These interactions actually help the survival of multiple myeloma cells and lead to difficulties with treatment. In the context of CAR-T therapy, the cytokines present in the TME prevent effective CAR-T cell infiltration and alter their ability to locate the desired target cells. These issues are further compounded by the short-term persistence of T cells following delivery to the patient, which limits the durability of the therapy and requires complete eradication of the target in a brief timespan to avoid remission.
The concerns regarding short-term persistence begin with the identification and optimization of the T cells’ sub-phenotypes, which are delivered to the patient following ex vivo expansion. Post-treatment phenotype analyses of cells delivered to responders vs. non-responders have highlighted that patient outcomes tend to be more favorable when there are higher percentages of memory (CD62L+ CCR7+) cells and a more equitable distribution of CD4 and CD8 cells [19][20]. However, most standard expansion systems used in CAR T cell manufacturing tend to simply target CD3 and CD28 in non-physiologically relevant manners, which tends to disproportionately expand CD8 effector cells [21]. While novel expansion systems are being developed with the goal of improving the phenotypic distribution of the cell product, their implementation in approved therapies would require new trials to formally validate their safety and efficacy [22][23].
Another consideration that affects the quality of the downstream product is the fact that current approval for CAR-T is restricted to cases of relapsed or refractory disease, which results in reduced cell fitness of the starting material and, therefore, a reduced fitness of the final cell product [9]. The advanced disease state of patients eligible for the therapy also causes problems with current manufacturing timelines, where vein-to-vein times could be as long as 4 weeks, depending on the product and the necessary quality control [4]. Further concerns involving the manufacturing of CAR-T cells regard the use of retroviral/lentiviral gene delivery for CAR expression, which can result in variable surface expression or issues with the randomness of the genetic insertion [24]. Additionally, vector fabrication tends to be a time-intensive and expensive process, particularly when considering the need to use larger scales as more therapies are approved [25]. There is some work on the development of mRNA-transduced CAR-T cells that lose expression of the receptor within 14 days to limit adverse effects but this remains in pre-clinical stages and requires extensive validation as a large-scale and durable alternative to viral transduction [26]. Finally, the cost to patients and insurers is a huge hurdle affecting the adoption of CAR-T therapy, given that the average total costs for administration easily exceed $400,000 and can climb as high as $1,000,000 [27].
6. Overlook
Researchers are cognizant of the current limitations of CAR-T and plenty of work is being carried out to improve the efficacy and quality of the therapy while expanding its accessibility. One approach to address the threat of antigen escape is to develop dual targeting, such as BCMA and transmembrane activator and CAML interactor (TACI), in the context of multiple myeloma using dual-binding ligands (APRILs), which are then targeted by the CAR [28]. Similarly, the co-expression of anti-BCMA and anti-GPRC5D CARs on the same cell has shown promising results in pre-clinical models of disease relapse [29]. Additionally, logic circuits can be introduced into CAR-T cells using synNotch receptors, which trigger transcriptional expression of the CAR molecule itself upon detection of an initial target [30]. These approaches allow for a more robust detection of multiple targeted surface markers, which increase the specificity of the therapy and should decrease the likelihood of exhaustion post-delivery. Other approaches to addressing exhaustion are the combinatorial delivery of CAR-T cells, along with immune checkpoint blockade or even the co-expression of CAR and IL-7 receptor [31][32].
Additionally, there is a push towards the development of techniques to remove HLA and TCR hurdles, which would cause graft-versus-host-disease (GvHD) in cases where non-donor matched CAR-T cells are delivered [33]. Developing allogeneic CAR-T cells that could be delivered to a wider array of patients would help to revolutionize the implementation of therapy by dramatically reducing its cost, due to the ability to scale-up, and reduce vein to vein times by having the therapy ready off-the-shelf. Finally, there is increased collaboration between industry and academia in order to proactively address manufacturing concerns. One of the largest and most visible collaborations is the NSF-funded Engineering Research Center for Cell Manufacturing Technologies (CMaT), whose work is focused on developing tools, assays, and supply chain solutions to improve the manufacture of cell therapeutics by working with industry and regulatory bodies to ensure facilitated transfer. These kinds of public–private partnerships will aid the alleviation of the financial burden of technology development for industry members—which, in turn, is relayed to patients—while still developing new methods to make these cutting-edge therapies more effective, durable, and cost-effective for the patients.
References
- Sadelain, M.; Brentjens, R.; Rivière, I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov. 2013, 3, 388–398.
- Benmebarek, M.-R.; Karches, C.; Cadilha, B.; Lesch, S.; Endres, S.; Kobold, S. Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. Int. J. Mol. Sci. 2019, 20, 1283.
- Hong, L.K.; Chen, Y.; Smith, C.C.; Montgomery, S.A.; Vincent, B.G.; Dotti, G.; Savoldo, B. CD30-Redirected Chimeric Antigen Receptor T Cells Target CD30+ and CD30− Embryonal Carcinoma via Antigen-Dependent and Fas/FasL Interactions. Cancer Immunol. Res. 2018, 6, 1274–1287.
- Fesnak, A.D.; June, C.H.; Levine, B.L. Engineered T cells: The promise and challenges of cancer immunotherapy. Nat. Rev. Cancer 2016, 16, 566–581.
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5, 177ra138.
- Carpenter, R.O.; Evbuomwan, M.O.; Pittaluga, S.; Rose, J.J.; Raffeld, M.; Yang, S.; Gress, R.E.; Hakim, F.T.; Kochenderfer, J.N. B-cell Maturation Antigen Is a Promising Target for Adoptive T-cell Therapy of Multiple Myeloma. Clin. Cancer Res. 2013, 19, 2048–2060.
- O’Connor, B.P.; Raman, V.S.; Erickson, L.D.; Cook, W.J.; Weaver, L.K.; Ahonen, C.; Lin, L.L.; Mantchev, G.T.; Bram, R.J.; Noelle, R.J. BCMA is essential for the survival of long-lived bone marrow plasma cells. J. Exp. Med. 2004, 199, 91–98.
- Rapoport, A.P.; Stadtmauer, E.A.; Binder-Scholl, G.K.; Goloubeva, O.; Vogl, D.T.; Lacey, S.F.; Badros, A.Z.; Garfall, A.; Weiss, B.; Finklestein, J.; et al. NY-ESO-1–specific TCR–engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 2015, 21, 914–921.
- Garfall, A.L.; Maus, M.V.; Hwang, W.-T.; Lacey, S.F.; Mahnke, Y.D.; Melenhorst, J.J.; Zheng, Z.; Vogl, D.T.; Cohen, A.D.; Weiss, B.M.; et al. Chimeric Antigen Receptor T Cells against CD19 for Multiple Myeloma. N. Engl. J. Med. 2015, 373, 1040–1047.
- Smith, E.L.; Harrington, K.; Staehr, M.; Masakayan, R.; Jones, J.; Long, T.J.; Ng, K.Y.; Ghoddusi, M.; Purdon, T.J.; Wang, X.; et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci. Transl. Med. 2019, 11, eaau7746.
- D’Agostino, M.; Raje, N. Anti-BCMA CAR T-cell therapy in multiple myeloma: Can we do better? Leukemia 2020, 34, 21–34.
- Teoh, P.J.; Chng, W.J. CAR T-cell therapy in multiple myeloma: More room for improvement. Blood Cancer J. 2021, 11, 1–18.
- Yin, Y.; Boesteanu, A.C.; Binder, Z.A.; Xu, C.; Reid, R.A.; Rodriguez, J.L.; Cook, D.R.; Thokala, R.; Blouch, K.; McGettigan-Croce, B.; et al. Checkpoint Blockade Reverses Anergy in IL-13Rα2 Humanized scFv-Based CAR T Cells to Treat Murine and Canine Gliomas. Mol. Ther. Oncolytics 2018, 11, 20–38.
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219–1226.
- Green, D.J.; Pont, M.; Sather, B.D.; Cowan, A.J.; Turtle, C.J.; Till, B.G.; Nagengast, A.M.; Libby, E.N., III; Becker, P.S.; Coffey, D.G.; et al. Fully Human Bcma Targeted Chimeric Antigen Receptor T Cells Administered in a Defined Composition Demonstrate Potency at Low Doses in Advanced Stage High Risk Multiple Myeloma. Blood 2018, 132, 1011.
- Samur, M.K.; Fulciniti, M.; Aktas Samur, A.; Bazarbachi, A.H.; Tai, Y.-T.; Prabhala, R.; Alonso, A.; Sperling, A.S.; Campbell, T.; Petrocca, F.; et al. Biallelic loss of BCMA as a resistance mechanism to CAR T cell therapy in a patient with multiple myeloma. Nat. Commun. 2021, 12, 1–7.
- Larson, R.C.; Maus, M.V. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat. Rev. Cancer 2021, 21, 145–161.
- Manier, S.; Sacco, A.; Leleu, X.; Ghobrial, I.M.; Roccaro, A.M. Bone Marrow Microenvironment in Multiple Myeloma Progression. J. Biomed. Biotechnol. 2012, 2012, 1–5.
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.-T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571.
- Wang, D.; Aguilar, B.; Starr, R.; Alizadeh, D.; Brito, A.; Sarkissian, A.; Ostberg, J.R.; Forman, S.J.; Brown, C.E. Glioblastoma-targeted CD4+ CAR T cells mediate superior antitumor activity. JCI Insight 2018, 3, e99048.
- McLellan, A.D.; Ali Hosseini Rad, S.M. Chimeric antigen receptor T cell persistence and memory cell formation. Immunol. Cell Biol. 2019, 97, 664–674.
- Dwarshuis, N.J.; Song, H.W.; Patel, A.; Kotanchek, T.; Roy, K. Functionalized microcarriers improve T cell manufacturing by facilitating migratory memory T cell production and increasing CD4/CD8 ratio. bioRxiv 2019, 646760.
- Zhang, D.K.Y.; Cheung, A.S.; Mooney, D.J. Activation and expansion of human T cells using artificial antigen-presenting cell scaffolds. Nat. Protoc. 2020, 15, 773–798.
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; Van Der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117.
- Morenweiser, R. Downstream processing of viral vectors and vaccines. Gene Ther. 2005, 12, S103–S110.
- Lin, L.; Cho, S.-F.; Xing, L.; Wen, K.; Li, Y.; Yu, T.; Hsieh, P.A.; Chen, H.; Kurtoglu, M.; Zhang, Y.; et al. Preclinical evaluation of CD8+ anti-BCMA mRNA CAR T cells for treatment of multiple myeloma. Leukemia 2021, 35, 752–763.
- Lyman, G.H.; Nguyen, A.; Snyder, S.; Gitlin, M.; Chung, K.C. Economic Evaluation of Chimeric Antigen Receptor T-Cell Therapy by Site of Care Among Patients With Relapsed or Refractory Large B-Cell Lymphoma. JAMA Netw. Open 2020, 3, e202072.
- Lee, L.; Draper, B.; Chaplin, N.; Philip, B.; Chin, M.; Galas-Filipowicz, D.; Onuoha, S.; Thomas, S.; Baldan, V.; Bughda, R.; et al. An APRIL-based chimeric antigen receptor for dual targeting of BCMA and TACI in multiple myeloma. Blood 2018, 131, 746–758.
- Fernández De Larrea, C.; Staehr, M.; Lopez, A.V.; Ng, K.Y.; Chen, Y.; Godfrey, W.D.; Purdon, T.J.; Ponomarev, V.; Wendel, H.-G.; Brentjens, R.J.; et al. Defining an Optimal Dual-Targeted CAR T-cell Therapy Approach Simultaneously Targeting BCMA and GPRC5D to Prevent BCMA Escape–Driven Relapse in Multiple Myeloma. Blood Cancer Discov. 2020, 1, 146–154.
- Choe, J.H.; Watchmaker, P.B.; Simic, M.S.; Gilbert, R.D.; Li, A.W.; Krasnow, N.A.; Downey, K.M.; Yu, W.; Carrera, D.A.; Celli, A.; et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci. Transl. Med. 2021, 13, eabe7378.
- Shum, T.; Omer, B.; Tashiro, H.; Kruse, R.L.; Wagner, D.L.; Parikh, K.; Yi, Z.; Sauer, T.; Liu, D.; Parihar, R.; et al. Constitutive Signaling from an Engineered IL7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer Discov. 2017, 7, 1238–1247.
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144.
- Qasim, W.; Amrolia, P.J.; Samarasinghe, S.; Ghorashian, S.; Zhan, H.; Stafford, S.; Butler, K.; Ahsan, G.; Gilmour, K.; Adams, S.; et al. First Clinical Application of Talen Engineered Universal CAR19 T Cells in B-ALL. Blood 2015, 126, 2046.
More
Information
Subjects:
Oncology; Immunology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
991
Revisions:
3 times
(View History)
Update Date:
03 Dec 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No