Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Andrea Henriques Pons | + 3302 word(s) | 3302 | 2021-11-19 07:01:13 | | | |
| 2 | Catherine Yang | + 8 word(s) | 3310 | 2021-11-22 02:56:07 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Henriques Pons, A. Muscular Dystrophies (MD). Encyclopedia. Available online: https://encyclopedia.pub/entry/16212 (accessed on 27 July 2026).
Henriques Pons A. Muscular Dystrophies (MD). Encyclopedia. Available at: https://encyclopedia.pub/entry/16212. Accessed July 27, 2026.
Henriques Pons, Andrea. "Muscular Dystrophies (MD)" Encyclopedia, https://encyclopedia.pub/entry/16212 (accessed July 27, 2026).
Henriques Pons, A. (2021, November 19). Muscular Dystrophies (MD). In Encyclopedia. https://encyclopedia.pub/entry/16212
Henriques Pons, Andrea. "Muscular Dystrophies (MD)." Encyclopedia. Web. 19 November, 2021.
Copy Citation
Muscular dystrophies (MDs) are genetic disorders caused by mutations in several genes that lead to the lack of or dysfunctional production of proteins that are essential for myofiber integrity and contraction. MDs are a group of diseases that cause, but are not restricted to, progressive muscle destruction and weakness, with nine most common forms: myotonic, Duchenne, Becker, limb-girdle, facioscapulohumeral, congenital, oculopharyngeal, distal, and Emery–Dreifuss.
muscular dystrophy
inflammation
genetic disorders
1. Myotonic Muscular Dystrophy (MMD)
MMD is a dominant and autosomal disease with two similar but distinct forms, type 1 (MMD1) and 2 (MMD2). MMD1 is also named Steinert’s Disease, in honor of the scientist who first described the disease in 1909. MMD1 is the most common form of the disease, and although most commonly found in adults, a late-onset is not a rule. The symptoms include muscle weakness and wasting, cardiac conduction defects, myotonia, diabetes and insulin resistance, cataracts, and many others, leading to a multisystem involvement. MMD1 can be divided into four different forms according to the clinical phenotype, illustrating the broad range of symptoms and general characteristics [1]. In adult-onset MMD, diagnostic efforts are usually initiated because of muscle weakness, myotonia, or cataracts—the three main symptoms. In this case, a family history of type 1 MMD combined with minor signs is a common starting point in the diagnostic examination. The progression of the disease is slow and advances to deepened skeletal muscle weakness, including muscles in the face, neck, and distal limbs. It progresses until eventual immobility, respiratory insufficiency, dysarthria, and dysphagia occur. The latter is one of the leading causes of severe disability and death in the late stages of adult-onset MMD1. The cardiac muscle shows conduction defects and tachyarrhythmia, most likely due to fibrosis. Usually, the detection of cataracts in older patients does not initiate further diagnostic considerations for MMD1. Still, it should particularly alert clinicians when detected in patients under 50 and associated with specific structural characteristics [2]. The central nervous system can also be affected, leading to progressive cognitive impairment and late apathy [3], besides gastrointestinal disorders such as constipation, incontinence, and diarrhea [4]. Endocrine dysfunction is also found, leading mostly to insulin resistance and susceptibility to diabetes, hypothyroidism, male hypogonadism, and adrenal insufficiency [5]. The second form of MMD1 is Congenital MMD; this is the most severe form of MMD, and is usually detected prenatally because of reduced fetal movements and different deformities. Babies have severe hypotonia in their limb, trunk, respiratory, facial, and bulbar muscles at birth, leading to respiratory dysfunction and feeding difficulties [6]. Mental retardation can also be observed. In childhood-onset MMD, there is no myotonia or muscle weakness, which imposes challenges for MD diagnosis and correct assistance. Instead, children have delayed learning at school and show signs of mental retardation. These patients also develop muscle weakness and wasting at an older age, causing physical disabilities comparable with severe adult-onset type 1 disease [7]. Finally, in late-onset oligosymptomatic MMD, the genetic family history is significant for clinicians considering an MMD diagnosis, as mild symptoms can be observed in earlier generations, like cataracts and discrete muscle weakness. However, later generations can have severe disease characteristics, including muscle wasting and atrophy, cataracts, and others [1].
The broad spectrum of tissues and organs affected, not restricted solely to muscles, and the disease severity in MMD1 is determined by the underlying molecular pathogenesis mediated by deleterious nuclear RNA repeats. Therefore, abnormalities in many pathways of RNA metabolism, including alternative splicing, can be detected in MMD1. In healthy cells, pre-mRNA splicing is mediated by nuclear protein factors that regulate the processing of mature RNA and the translation of functional proteins. In healthy individuals, there are low numbers of CTG repeat in the 3′-UTR untranslated region of the dystrophia myotonica protein kinase (DMPK) gene (Figure 1A). However, in MMD1, there is an unstable expansion of this repeat [8], leading to DMPK transcripts that have expanded CUG repeats (CUGnDMPK). These structures are retained and distributed in the cells’ nuclei as discrete and visible foci. The repeats sequester double-stranded CUG-binding proteins, including the best-characterized player in this mechanism, the muscleblind-like (MBNL) family of splicing factors (Figure 1B). These proteins interact physically with expanded CUG repeats of the DMPK RNA and drastically reduce its availability for unprocessed RNA strains. One of the main pre mRNA targets of the CUGnDMPK + MBNL is the chloride channel RNA construct (CLCN1), which leads to aberrantly spliced CLCN1 transcripts with additional exons and premature termination codons. Then, there is cytoplasmic degradation through the nonsense-mediated decay pathway, truncated nonfunctional CLCN1 protein, and/or dysfunctional channel activity [9] (Figure 1B). All these alterations lead to the increased muscle excitability observed in MMD patients.
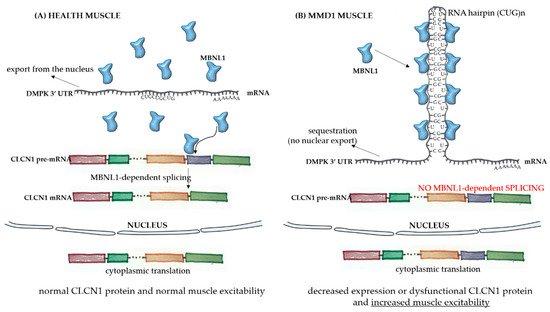
Figure 1. Pathophysiology of myotonic muscular dystrophy type 1. In healthy skeletal muscle fibers (A), the mRNA sequence of the dystrophia myotonica protein kinase (DMPK) gene has a limited number of the repeat sequence CUG, and there is normal export of RNA to the cytoplasm. The MBNL1 factor is available to play its role in processing other transcripts, such as the pre mRNA of CLCN1, in the nucleus. After the correct splicing, mature mRNAs are exported to the cytoplasm for protein translation and normal function in subcellular compartments. In the case of muscle fibers from myotonic muscular dystrophy type 1 (MMD1) patients (B), there is an aberrant expansion of the CUG repeat in the DMPK mRNA, with the formation of a hairpin due to the pairing of the bases composing the repeats. In this case, there is the sequestration of the MBNL1 factor, which binds to the hairpin. The complex then precipitates into microscopically visible nuclear foci. Because of the MBNL1 retention, the nuclear splicing of pre mRNAs is compromised, leading to decreased or dysfunctional protein production.
It is known that the number of repeated CUG sequences is directly related to disease severity, with 38 to 50 repeat sizes being considered premutations and generally not leading to apparent symptoms. On the other hand, mild phenotypes are associated with sequences with 51 to 149 repeats, and early, more severe onset phenotypes have more extended repeat sequences, with at least 1000 repeats [10]. The distribution of affected tissues and organs is at least partially explained by somatic mosaicism due to repeat size instability during mitosis. Peripheral blood leukocytes, for example, have smaller expansion sizes when compared with other cell types and cells from other tissues [10].
Although there are many overlapping phenotypic features in both types of MMD, key characteristics that distinguish MMD2 from MMD1 include more proximal muscle weakness and general milder cardiac and multisystem involvement in MMD2 [11]. In contrast to MMD1, MMD2 onset occurs in adulthood, with no reports of congenital development. In MMD2, there is a (CCTG)n expansion in intron 1 of the cellular nucleic acid-binding protein/zinc finger protein 9 (CNBP/ZNF9) [12] and sequestration of MBNL in the hairpin of repeats [13]. Still, the pathophysiology of MMD2 is not well understood [14].
There is no cure for MMD, but studies of new therapeutic strategies focus primarily on reducing RNA repeat sequences and preventing interactions of the RNA hairpins of repeated sequences with MBNL. Antisense oligonucleotides for the CUG repeats in the DMPK transcripts are especially promising, potentially discriminating between normal and mutated transcripts.
2. Oculopharyngeal Muscular Dystrophy (OPMD)
OPMD is a late-onset neuromuscular disorder that starts with lowering the eyelids (ptosis) and swallowing difficulties (dysphagia). With the progression of the disease, other skeletal muscles can be affected, including the proximal muscles of the lower limbs. In general, the symptoms of OPMD are similar to myasthenia gravis, for example, and clinicians must pay special attention to some pathological hallmarks, like the formation of insoluble inclusions in the nuclei of muscle cells [15]. OPMD is a monogenic disorder, with a mutation in the gene encoding for poly-adenylate (poly(A)) binding protein nuclear 1 (PABPN1) [16], leading to a short GCG expansion in its polyalanine tract. This protein is a multifunctional regulator of RNA metabolism [17][18][19]. Despite its ubiquitous expression, mutated PABPN1 leads to symptoms manifested predominantly in skeletal muscles, where the protein expression levels are lower [20]. It is unclear why the disease initiates from midlife onward, nor is the precise nature of the correlation between mutated PABPN1 levels and the poly(A) tail length, or how the protein regulates changes in the RNA metabolism. Although some molecular therapies are under investigation [21][22], current treatments for OPMD are limited to surgical corrections for ptosis and dysphagia, targeting the cricopharyngeal muscle.
3. Emery–Dreifuss Muscular Dystrophy (EDMD)
EDMD was characterized by its clinical features and disease course in 1966 [23]. It mainly affects the brachial and fibular muscle groups and induces multiarticular contractures and spine rigidity, besides cardiomyopathy with conduction disturbances later [24][25][26]. The first symptoms appear in the first decade of life, manifesting as ankle and elbow contractures and spine rigidity. In the second and third decades of life, muscle atrophy and weakness are more visible, usually with a slow progression [27]. EDMD is divided into at least four types (1 to 4), and EDMD1 is associated with mutations in the emerin gene (EMD) located on the X chromosome. This protein spans the inner nuclear membrane and regulates several nuclear functions (Figure 2). Emerin binds to lamins A and C to form the nuclear lamina, a proteinaceous network that regulates the architecture and function of nuclear DNA. The emerin protein regulates gene expression by binding to and regulating the activity of many transcription factors and downstream signaling pathways, including the histone deacetylase 3 (HDAC3). This enzyme alters the chromatin structure and regulates transcription factors activity. The EMD gene contains six exons, and the first mutation associated with EDMD was identified as c.3G > A, affecting codon start in exon 1. Since then, the nonsense mutation c.130C > T in exon 2 and c.653insTGGGC in exon 6 have also been identified, influencing an open reading frame that creates a premature stop codon at the 238 position [28]. In most EDMD1 cases, small deletions or splice-site mutations lead to this codon change, while the remaining patients have nonsense/missense mutations or large deletions [29].
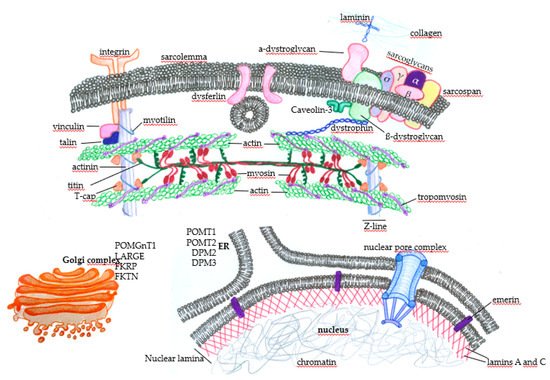
Figure 2. Subcellular distribution of some proteins that, when genetically altered, lead to muscular dystrophies. In the sarcolemma, there are proteins, such as the components of the dystrophin-associated glycoprotein complex (DGC) (α-dystroglycan, σ, γ, β, and α sarcoglycans, sarcospan, and β-dystroglycan), that physically mediate the interaction of the extracellular ambient with the cytoplasm. It happens through the binding of laminin with extracellular matrix components such as collagen. The protein dystrophin mediates the integration of the complex with actin filaments, composing the contractile structures of the muscle cells’ cytoskeleton. The concerted action of these proteins protects the muscle fibers against cellular damage during the contraction cycles. Membrane integrins also interact with extracellular matrix components and provide normal muscle function and contraction by integrating with intracellular components. Under physiological circumstances, when there are minor disruptions of the sarcolemma, the dysferlin mediates membrane repair to avoid cell death. Many proteins play essential roles in maintaining normal muscle function and contraction force, such as vinculin, talin, and myotilin, alongside titin, T-cap (composing the Z line), tropomyosin, and the main contractile components, myosin and actin. Nuclear proteins such as emerin, which anchors the nuclear lamina components lamin A and C, can also induce muscular dystrophies when genetically mutated. Finally, alterations in the glycosylation patterns of proteins such as (protein o-mannosyltransferase) POMT1, POMT2, (dolichol-phosphate-mannose) DPM2, and DPM3 in the endoplasmic reticulum (ER) and (protein O-mannose b-1, 2-N-acetylglucosaminyltransferase) POMGnT1, (like-glycosyltransferase) LARGE, (Fukutin-related protein) FKRP, and (Fukutin) FKTN in the Golgi complex deeply affect muscle function and lead to muscular dystrophy.
In EDMD2, symptom severity varies extensively, with muscle atrophy, joint contractures, and loss of ambulance occurring early in some patients, in contrast to patients with mild and late-onset, which are associated with slow progression [30]. Respiratory muscle weakness and chest deformities usually lead to respiratory failure, while left ventricle systolic dysfunction and cardiac conduction disturbances lead to dilated cardiomyopathy and sudden death. The autosomic LNMA gene has been identified to be responsible for EDMD2; it is located in the long arm of chromosome 1 at position 22(1q22) and encodes the lamins, mainly lamin A and C (Figure 2). Genetic alterations involve heterozygous missense mutations in most patients and generate lamins A [31] and C with a dominant-negative toxic effect. However, some deletions or duplications may lead to not functional proteins [32].
Identifying other genes related to the organization of nuclear structures that interface with the chromatin has led to the description of different types of EDMD. For example, mutations in the SYNE1 and SYNE2 genes (the synaptic nuclear envelope protein 1(or 2)), encoding for nesprin-1 and nesprin-2, are linked to EDMD4 and EDMD5, respectively [33]. Another example is the LAP gene, which encodes polypeptides connected with lamins (lamin-associated protein 2 alpha, LAP2alpha) [34] and mutations in the FHL-1 (four and a half LIM domains protein 1) gene located on the X chromosome [35]. Unfortunately, EDMD treatment also relies primarily on targeting the symptoms, including surgical procedures.
4. Limb-Girdle Muscular Dystrophy (LGMD)
LGMDs are a highly heterogeneous group of autosomal dominant or recessive disorders that, in general terms, lead to weakness and wasting of muscles in the legs and arms. LGMDs are usually not syndromic and are mostly limited to skeletal muscles. However, some patients have cardiac problems [36], and a few patients exhibit evolutionary delays and mental disabilities [37]. LGMD patients have highly variable phenotypes and clinical courses and can be affected at any age [38]; however, one of the few unifying characteristics is that the symptoms are more severe when the disease manifests early in life [39]. The phenotypic heterogeneity reflects the multiple genes involved in LGMD—genes involved in muscle fiber integrity, repair, and function, producing abnormal proteins in the sarcomere, sarcolemma extracellular matrix, nucleus, and other cell parts [40].
Taghizadeh [41] published that more than 30 different subtypes of LGMD have been described so far. The most common subtypes are LGMD1A (myotilinopathy), LGMD1B (laminopathy), LGMD1C (caveolinopathy), LGMD1E and LGMD2R (desminopathies), LGMD2A (calpainopathy), LGMD2B (dysferlinopathy), LGMD2C, 2D, 2E, and 2F (the sarcoglycanopathies), LGMD2G (telethoninopathy), LGMD2I, 2K, 2M, 2N, 2O, 2P, 2T, and 2U (the dystroglycanopathies), LGMD2J (titinopathy), and LGMD2L. Figure 2 summarizes some of the LGMD-associated proteins and illustrates the wide range of muscular functions affected by mutations in the disease.
5. Facioscapulohumeral Muscular Dystrophy (FSHD)
FSHD is one of the most frequent MDs and affects mainly the muscles of the face, shoulder blades, and upper arms, but muscle weakness is usually progressively observed in other muscles. The first signs start before the 20s, with weakness and discrete atrophy of the muscles around the mouth and eyes, and abdominal and hip muscles can be affected later. FSHD progresses very slowly, rarely affecting the cardiac or respiratory system, and most patients have an average life span [42][43].
Most FSHD type 1 patients have a genetic mutation leading to the pathogenic loss of D4Z4 microsatellite repeats [44], and each D4Z4 unit on chromosome 4q35 contains a copy of the DUX4 (double homeobox 4) retrogene, a germline transcription factor [8]. In healthy individuals, 11 to 100 D4Z4 repeats (each repeat with 3.3 kb) are observed, with normal chromatin methylation in most cases and no transcription of DUX4 in somatic tissues. However, FSHD patients have a contraction in the number of repeats, varying from 1 to 10, with a reduction in the number of repeats associated with disease severity. The presence of at least one repeat containing a copy of the DUX4 gene is required for FSHD development [45], leading to hypomethylation in most cases and DUX4 transcription (decreased repression of the D4Z4 repeat). However, it seems that the transcribed full-length DUX4 mRNA is unstable due to the lack of a polyadenylation signal sequence, and much remains to be understood about the pathophysiology underlying the FSHD phenotype [46]. Moreover, mutations in the genes SMCHD1 or DNMT3B are associated with FSHD type 2, also leading to aberrant expression of DUX4 in skeletal muscles and similar symptoms to FSHD type 1.
6. Congenital Muscular Dystrophy (CMD) and Distal Muscular Dystrophy
CMDs are also a highly heterogeneous group of very early-onset severe muscle diseases with symptoms that include decreased mobility, delay or arrest of gross motor development, and joint or spinal deformities that impose significant difficulties on patients’ development. With the progression of the disease, cardiac and respiratory complications may occur and, in some subtypes, the central nervous system and connective tissue can be affected [47][48][49]. A final diagnosis of CMD requires an integrated clinical and pathological analysis [37] because of a relatively overlapping spectrum of symptoms spanning characteristics of congenital myopathies, congenital myasthenic syndromes, LGMD, and others [47]. There are more than 13 genes associated with CMD, and the primary disease subtypes are caused by laminin alpha-2 (merosin) deficiency (MDC type 1A, MDC1A) or partial merosin deficiency (MDC1B), fukutin-related proteinopathy (MDC1C), or acetylglucosaminyltransferase-like protein (LARGE)-related CMD (MCD1D) [50]. Other affected proteins include collagen VI, integrin α7, selenoprotein N, fukutin, O-mannosyltransferase 1 (POMT1) (Figure 2), and many others, generating a complex disease from the perspective of diagnosis and treatment.
Distal myopathies are also clinically, pathologically, and genetically heterogeneous, with highly variable phenotypes. In contrast to CMD, the onset age ranges from childhood to late adulthood, and muscle weakness usually starts at very distal muscles, like the finger and toe extensor muscles. As the disease progresses, proximal muscles may become impacted, but the distal weakness remains the most severe. Most distal myopathies are genetically determined, although acquired myopathies can occasionally manifest with distal weakness (e.g., sporadic inclusion body myositis, sarcoid myopathy, or focal myositis [51]). Mutations in many different genes encoding proteins such as caveolin-3, dysferlin, α-actin-1, myotilin, desmin, and many others also lead to distal muscular dystrophy.
7. Duchenne Muscular Dystrophy (DMD) and Becker Muscular Dystrophy (BMD)
DMD is an X-linked disorder and the most common form of MD, affecting one in 3600 to 6000 male births. It is characterized by progressive muscle wasting, with disease onset around three years of age and patients becoming wheelchair users around their teens. Cardiorespiratory complications become progressively more severe, and death usually occurs in the second decade of life. Proximal muscle groups are the most affected muscles, leading to Gower’s sign when the child climbs up on their own body to reach an upright posture. Calf enlargement is also observed as a sign of muscle inflammation and fat deposition [52][53]. Increased blood CK and hepatic transaminase levels combined with specific clinical signs and biopsy analysis or DNA testing are used to make a diagnosis of DMD [53]. DMD is caused by mutations, mainly deletions, altering the open reading frames of the dystrophin gene, which has 79 exons, resulting in the absence of dystrophin [54]. This intracellular protein connects the cytoskeleton to the DGC, linking the intracellular and extracellular environments (Figure 2). The lack of dystrophin leads to sarcolemma disruption, mainly due to mechanical damage and local inflammation. With continuous cycles of damage and repair, the myogenic cells become exhausted, and muscle regeneration is compromised. In later stages of the disease, the accumulation of fibrosis further deteriorates muscle function [55]. There is no cure for DMD, and patient management is multidisciplinary, involving general and respiratory physiotherapy, genetic counseling, and corticosteroid treatment. This pharmacological agent is currently used to improve muscle strength, cardiorespiratory function, and life quality and expectancy. Despite its efficacy, corticosteroid-based therapy has many side effects that are detrimental to patients [56]. Several new therapy proposals are emerging, aiming to improve the life span of people with DMD [57]. These new strategies aim to genetically restore dystrophin expression in vivo [58] to stabilize the sarcolemma and enhance the expression of compensatory proteins, such as utrophin [59][60].
BMD is a mild phenotype of dystrophinopathy, with about one-third of the incidence of DMD. The underlying pathological difference between the two diseases is that the mutations in the dystrophin gene of BMD do not abort the protein’s expression but partially affect its function, leading to a milder clinical phenotype. BMD patients usually show symptoms later, around 12 years of age, with a higher incidence of cardiac dysfunction and death typically around the fourth decade of life [61][62].
The broad spectrum of pathogenic mutations and clinical symptoms associated with no curative therapies reinforce the need to search for new treatments for MD patients. Stem-cell-based therapies are a promising alternative for most of these patients, and many efforts have been made to overcome the numerous limitations of these procedures.
References
- Udd, B.; Krahe, R. The myotonic dystrophies: Molecular, clinical, and therapeutic challenges. Lancet Neurol. 2012, 11, 891–905.
- Garrott, H.M.; Walland, M.J.; O’Day, J. Recurrent posterior capsular opacification and capsulorhexis contracture after cataract surgery in myotonic dystrophy. Clin. Exp. Ophthalmol. 2004, 32, 653–655.
- Winblad, S.; Lindberg, C.; Hansen, S. Temperament and character in patients with classical myotonic dystrophy type 1 (DM-1). Neuromuscul. Disord. 2005, 15, 287–292.
- Rönnblom, A.; Forsberg, H.; Danielsson, A. Gastrointestinal symptoms in myotonic dystrophy. Scand. J. Gastroenterol. 1996, 31, 654–657.
- Takeshima, K.; Ariyasu, H.; Ishibashi, T.; Kawai, S.; Uraki, S.; Koh, J.; Ito, H.; Akamizu, T. Myotonic dystrophy type 1 with diabetes mellitus, mixed hypogonadism and adrenal insufficiency. Endocrinol. Diabetes. Metab. Case Rep. 2018, 2018.
- Ashizawa, T.; Sarkar, P.S. Myotonic dystrophy types 1 and 2. Handb. Clin. Neurol. 2011, 101, 193–237.
- Angeard, N.; Huerta, E.; Jacquette, A.; Cohen, D.; Xavier, J.; Gargiulo, M.; Servais, L.; Eymard, B.; Héron, D. Childhood-onset form of myotonic dystrophy type 1 and autism spectrum disorder: Is there comorbidity? Neuromuscul. Disord. 2018, 28, 216–221.
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.P.; Hudson, T. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3’ end of a transcript encoding a protein kinase family member. Cell 1992, 69, 385.
- López-Morató, M.; Brook, J.D.; Wojciechowska, M. Small Molecules Which Improve Pathogenesis of Myotonic Dystrophy Type 1. Front. Neurol. 2018, 9, 349.
- LoRusso, S.; Weiner, B.; Arnold, W.D. Myotonic Dystrophies: Targeting Therapies for Multisystem Disease. Neurotherapeutics 2018, 15, 872–884.
- Day, J.W.; Ricker, K.; Jacobsen, J.F.; Rasmussen, L.J.; Dick, K.A.; Kress, W.; Schneider, C.; Koch, M.C.; Beilman, G.J.; Harrison, A.R.; et al. Myotonic dystrophy type 2: Molecular, diagnostic and clinical spectrum. Neurology 2003, 60, 657–664.
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001, 293, 864–867.
- Schoser, B.; Timchenko, L. Myotonic dystrophies 1 and 2: Complex diseases with complex mechanisms. Curr. Genomics. 2010, 11, 77–90.
- Ranum, L.P.; Rasmussen, P.F.; Benzow, K.A.; Koob, M.D.; Day, J.W. Genetic mapping of a second myotonic dystrophy locus. Nat. Genet. 1998, 19, 196–198.
- Tomé, F.M.; Fardeau, M. Nuclear inclusions in oculopharyngeal dystrophy. Acta Neuropathol. 1980, 49, 85–87.
- Brais, B.; Bouchard, J.P.; Xie, Y.G.; Rochefort, D.L.; Chrétien, N.; Tomé, F.M.; Lafrenière, R.G.; Rommens, J.M.; Uyama, E.; Nohira, O.; et al. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat. Genet. 1998, 18, 164–167.
- Kerwitz, Y.; Kühn, U.; Lilie, H.; Knoth, A.; Scheuermann, T.; Friedrich, H.; Schwarz, E.; Wahle, E. Stimulation of poly(A) polymerase through a direct interaction with the nuclear poly(A) binding protein allosterically regulated by RNA. EMBO J. 2003, 22, 3705–3714.
- Benoit, B.; Mitou, G.; Chartier, A.; Temme, C.; Zaessinger, S.; Wahle, E.; Busseau, I.; Simonelig, M. An essential cytoplasmic function for the nuclear poly(A) binding protein, PABP2, in poly(A) tail length control and early development in Drosophila. Dev. Cell 2005, 9, 511–522.
- Bresson, S.M.; Conrad, N.K. The human nuclear poly(a)-binding protein promotes RNA hyperadenylation and decay. PLoS Genet. 2013, 9, e1003893.
- Malerba, A.; Klein, P.; Bachtarzi, H.; Jarmin, S.A.; Cordova, G.; Ferry, A.; Strings, V.; Espinoza, M.P.; Mamchaoui, K.; Blumen, S.C.; et al. PABPN1 gene therapy for oculopharyngeal muscular dystrophy. Nat. Commun. 2017, 8, 14848.
- Davies, J.E.; Berger, Z.; Rubinsztein, D.C. Oculopharyngeal muscular dystrophy: Potential therapies for an aggregate-associated disorder. Int. J. Biochem. Cell Biol. 2006, 38, 1457–1462.
- Barbezier, N.; Chartier, A.; Bidet, Y.; Buttstedt, A.; Voisset, C.; Galons, H.; Blondel, M.; Schwarz, E.; Simonelig, M. Antiprion drugs 6-aminophenanthridine and guanabenz reduce PABPN1 toxicity and aggregation in oculopharyngeal muscular dystrophy. EMBO Mol. Med. 2011, 3, 35–49.
- Emery, A.E.; Dreifuss, F.E. Unusual type of benign x-linked muscular dystrophy. J. Neurol. Neurosurg. Psychiatry 1966, 29, 338–342.
- Zacharias, A.S.; Wagener, M.E.; Warren, S.T.; Hopkins, L.C. Emery-Dreifuss muscular dystrophy. Semin Neurol 1999, 19, 67–79.
- Helbling-Leclerc, A.; Bonne, G.; Schwartz, K. Emery-Dreifuss muscular dystrophy. Eur. J. Hum. Genet. 2002, 10, 157–161.
- Mercuri, E.; Muntoni, F. Muscular dystrophies. Lancet 2013, 381, 845–860.
- Madej-Pilarczyk, A.; Kochański, A. Emery-Dreifuss muscular dystrophy: The most recognizable laminopathy. Folia Neuropathol. 2016, 54, 1–8.
- Bione, S.; Maestrini, E.; Rivella, S.; Mancini, M.; Regis, S.; Romeo, G.; Toniolo, D. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat. Genet. 1994, 8, 323–327.
- Manilal, S.; Recan, D.; Sewry, C.A.; Hoeltzenbein, M.; Llense, S.; Leturcq, F.; Deburgrave, N.; Barbot, J.; Man, N.; Muntoni, F.; et al. Mutations in Emery-Dreifuss muscular dystrophy and their effects on emerin protein expression. Hum. Mol. Genet. 1998, 7, 855–864.
- Bonne, G.; Mercuri, E.; Muchir, A.; Urtizberea, A.; Bécane, H.M.; Recan, D.; Merlini, L.; Wehnert, M.; Boor, R.; Reuner, U.; et al. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann. Neurol. 2000, 48, 170–180.
- Angori, S.; Capanni, C.; Faulkner, G.; Bean, C.; Boriani, G.; Lattanzi, G.; Cenni, V. Emery-Dreifuss Muscular Dystrophy-Associated Mutant Forms of Lamin A Recruit the Stress Responsive Protein Ankrd2 into the Nucleus, Affecting the Cellular Response to Oxidative Stress. Cell Physiol. Biochem. 2017, 42, 169–184.
- Benedetti, S.; Menditto, I.; Degano, M.; Rodolico, C.; Merlini, L.; D’Amico, A.; Palmucci, L.; Berardinelli, A.; Pegoraro, E.; Trevisan, C.P.; et al. Phenotypic clustering of lamin A/C mutations in neuromuscular patients. Neurology 2007, 69, 1285–1292.
- Zhang, Q.; Bethmann, C.; Worth, N.F.; Davies, J.D.; Wasner, C.; Feuer, A.; Ragnauth, C.D.; Yi, Q.; Mellad, J.A.; Warren, D.T.; et al. Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum. Mol. Genet. 2007, 16, 2816–2833.
- Gesson, K.; Vidak, S.; Foisner, R. Lamina-associated polypeptide (LAP)2α and nucleoplasmic lamins in adult stem cell regulation and disease. Semin. Cell Dev. Biol. 2014, 29, 116–124.
- Gueneau, L.; Bertrand, A.T.; Jais, J.P.; Salih, M.A.; Stojkovic, T.; Wehnert, M.; Hoeltzenbein, M.; Spuler, S.; Saitoh, S.; Verschueren, A.; et al. Mutations of the FHL1 gene cause Emery-Dreifuss muscular dystrophy. Am. J. Hum. Genet. 2009, 85, 338–353.
- Wahbi, K.; Meune, C.; Hamouda, E.H.; Stojkovic, T.; Laforêt, P.; Bécane, H.M.; Eymard, B.; Duboc, D. Cardiac assessment of limb-girdle muscular dystrophy 2I patients: An echography, Holter ECG and magnetic resonance imaging study. Neuromuscul. Disord. 2008, 18, 650–655.
- Bönnemann, C.G.; Wang, C.H.; Quijano-Roy, S.; Deconinck, N.; Bertini, E.; Ferreiro, A.; Muntoni, F.; Sewry, C.; Béroud, C.; Mathews, K.D.; et al. Diagnostic approach to the congenital muscular dystrophies. Neuromuscul. Disord. 2014, 24, 289–311.
- Vissing, J. Limb girdle muscular dystrophies: Classification, clinical spectrum and emerging therapies. Curr. Opin. Neurol. 2016, 29, 635–641.
- Guglieri, M.; Magri, F.; D’Angelo, M.G.; Prelle, A.; Morandi, L.; Rodolico, C.; Cagliani, R.; Mora, M.; Fortunato, F.; Bordoni, A.; et al. Clinical, molecular, and protein correlations in a large sample of genetically diagnosed Italian limb girdle muscular dystrophy patients. Hum. Mutat. 2008, 29, 258–266.
- Liewluck, T.; Milone, M. Untangling the complexity of limb-girdle muscular dystrophies. Muscle Nerve. 2018, 58, 167–177.
- Taghizadeh, E.; Rezaee, M.; Barreto, G.E.; Sahebkar, A. Prevalence, pathological mechanisms, and genetic basis of limb-girdle muscular dystrophies: A review. J. Cell Physiol. 2019, 234, 7874–7884.
- Padberg, G.W.; Lunt, P.W.; Koch, M.; Fardeau, M. Diagnostic criteria for facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 1991, 1, 231–234.
- Statland, J.M.; Tawil, R. Facioscapulohumeral Muscular Dystrophy. Continuum. Minneap. Minn. 2016, 22, 1916–1931.
- van Deutekom, J.C.; Wijmenga, C.; van Tienhoven, E.A.; Gruter, A.M.; Hewitt, J.E.; Padberg, G.W.; van Ommen, G.J.; Hofker, M.H.; Frants, R.R. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum. Mol. Genet. 1993, 2, 2037–2042.
- Hamel, J.; Tawil, R. Facioscapulohumeral Muscular Dystrophy: Update on Pathogenesis and Future Treatments. Neurotherapeutics 2018, 15, 863–871.
- Lemmers, R.J.; Tawil, R.; Petek, L.M.; Balog, J.; Block, G.J.; Santen, G.W.; Amell, A.M.; van der Vliet, P.J.; Almomani, R.; Straasheijm, K.R.; et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet. 2012, 44, 1370–1374.
- Fu, X.N.; Xiong, H. Genetic and Clinical Advances of Congenital Muscular Dystrophy. Chin. Med. J. Engl. 2017, 130, 2624–2631.
- Mercuri, E.; Muntoni, F. The ever-expanding spectrum of congenital muscular dystrophies. Ann. Neurol. 2012, 72, 9–17.
- Falsaperla, R.; Praticò, A.D.; Ruggieri, M.; Parano, E.; Rizzo, R.; Corsello, G.; Vitaliti, G.; Pavone, P. Congenital muscular dystrophy: From muscle to brain. Ital. J. Pediatr. 2016, 42, 78.
- Kang, P.B.; Morrison, L.; Iannaccone, S.T.; Graham, R.J.; Bönnemann, C.G.; Rutkowski, A.; Hornyak, J.; Wang, C.H.; North, K.; Oskoui, M.; et al. Evidence-based guideline summary: Evaluation, diagnosis, and management of congenital muscular dystrophy: Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology 2015, 84, 1369–1378.
- Pénisson-Besnier, I. Distal myopathies. Rev. Neurol. Paris 2013, 169, 534–545.
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010, 9, 77–93.
- Ryder, S.; Leadley, R.M.; Armstrong, N.; Westwood, M.; de Kock, S.; Butt, T.; Jain, M.; Kleijnen, J. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: An evidence review. Orphanet J. Rare Dis. 2017, 12, 79.
- Hoffman, E.P.; Brown, R.H.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928.
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Phys. Rev. 2002, 82, 291–329.
- Yiu, E.M.; Kornberg, A.J. Duchenne muscular dystrophy. J. Paediatr. Child. Health 2015, 51, 759–764.
- Nakamura, A. Mutation-Based Therapeutic Strategies for Duchenne Muscular Dystrophy: From Genetic Diagnosis to Therapy. J. Pers. Med. 2019, 9.
- Okada, T.; Takeda, S. Current Challenges and Future Directions in Recombinant AAV-Mediated Gene Therapy of Duchenne Muscular Dystrophy. Pharm. Basel 2013, 6, 813–836.
- Fairclough, R.J.; Wood, M.J.; Davies, K.E. Therapy for Duchenne muscular dystrophy: Renewed optimism from genetic approaches. Nat. Rev. Genet. 2013, 14, 373–378.
- Mah, J.K. Current and emerging treatment strategies for Duchenne muscular dystrophy. Neuropsychiatr. Dis. Treat. 2016, 12, 1795–1807.
- Flanigan, K.M. Duchenne and Becker muscular dystrophies. Neurol Clin. 2014, 32, 671–688.
- Flanigan, K.M. The muscular dystrophies. Semin Neurol 2012, 32, 255–263.
More
Information
Subjects:
Genetics & Heredity
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
795
Revisions:
2 times
(View History)
Update Date:
22 Nov 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No