 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Anastasiya Snezhkina | + 2403 word(s) | 2403 | 2021-11-08 09:57:58 | | | |
| 2 | Vivi Li | Meta information modification | 2403 | 2021-11-11 04:27:24 | | |
Video Upload Options
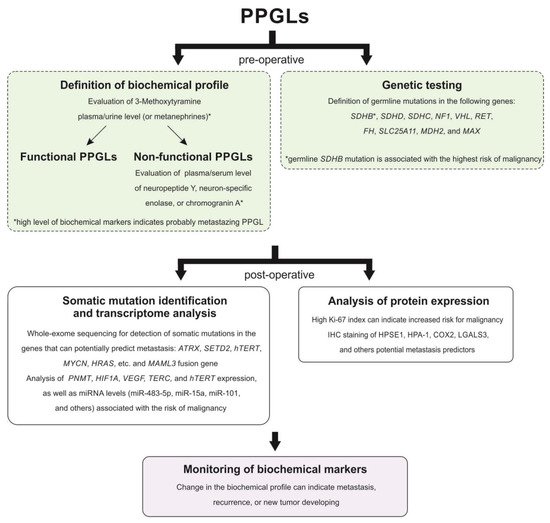
Paragangliomas and pheochromocytomas (PPGLs) are rare neuroendocrine tumors formed from paraganglionic tissue. Since 2017, PPGLs have been classified as tumors with variable potential to metastasize. Metastasizing PPGLs are usually difficult to diagnose and require evidence of regional or distant metastasis. Data on diagnostic and prognostic molecular markers for PPGL malignancy are limited, and many of the proposed factors remain controversial. There is a significant gap in the understanding of tumor pathogenesis, as well as the treatment and management of patients with PPGLs. This entry summarized the current findings on the potential markers for distinguishing between metastasizing and benign tumors, as well as on the prediction of aggressive behavior of PPGLs, especially of those localized in the head and neck region.
1. Definition, Localization, and Distribution
| Parameter | HNPGLs | PHEO | Other Extra-Adrenal PGLs | ||
|---|---|---|---|---|---|
| CPGL | MEPGL | VPGL | |||
| Mean age at diagnosis | 40–50 * [7][8] | 55 [3] | 41–47 [3] | 40–50 [9] | 40–50 [9] |
| Female/male ratio | 2:1–8:1 ** [10][11] | 3:1–9:1 [12] | 2:1–8:1 [12] | 1:1 [9] | 1:1 [9] |
| Multifocal cases, % | 10–25 [4] | 10–50 [13] | 10 *** [14] | 8 [15] | 33 [15] |
| Metastatic cases, % | 4–6 [12] | 2 [12] | 16–19 [10][12] | 10 [9] | 2.5–50 [9] |
2. Metastatic Disease
2.1. Clinical Characteristics
2.2. Biochemical Markers
2.3. Genetic Markers
Among the main genetic features associated with a high risk of the development of metastasizing PPGLs is a germline mutation in the SDHB gene. Testing for the germline SDHB mutation in patients with PPGLs is recommended by Clinical Practice Guidelines [26][27]. According to a systematic review and a meta-analysis study, the pooled incidence risk of metastasizing PPGLs for the SDHB mutation carriers was 17% while the prevalence ranged from 13% to 23% [51]. Among the patients with HNPGLs, the reported incidence of metastasis reaches 83% in the groups of SDHB mutation carriers.
The germline mutation in the SDHD gene was also reported in metastasizing PPGLs; however, the risk of metastasis development in SDHD mutation carriers is significantly lower than in those with an SDHB mutation [52]. SDHD mutations are more frequently associated with HNPGLs and multiple tumors [53][54][55]. The pooled risk of incidence and prevalence of metastasizing PPGLs for SDHD mutation carriers was estimated as 8% and 3%, respectively [51]. The incidence risk of malignancy for patients with SDHD-mutated HNPGLs reaches 22.7%. The highest incidence risk of malignancy (100%, 4/4) was observed among patients with HNPGLs from the Dutch population; at the same time, no variants were found in the SDHB gene [56]. All these patients carried a founder mutation in the SDHD gene. Thus, this higher association of the SDHD mutation with malignancy compared with SDHB, which was found in most studies, can be explained by characteristics of the Dutch population.
Several studies reported germline mutations in other susceptibility genes for PPGLs, such as FH [57], SLC25A11 [58], and MDH2 [59], which were associated with aggressive tumor behavior. These genes are classified as cluster 1 TCA cycle-related associated with the pseudohypoxia subtype of PPGLs [60]. Moreover, tumors with mutations in these genes were clustered together with SDHx-mutated tumors demonstrating similar hypermethylation profiles [61][58][59][62]. This phenotype seems to be involved in tumor progression and mutations in the FH, SLC25A11, and MDH2 genes along with SDHB and SDHD mutations can be considered to be a risk factor for PPGL malignancy. However, mutation frequency in these genes is rare and accounts for less than 1% [57][58][63]. Notably, alterations in FH and SLC25A11 were found in HNPGLs but in non-metastasizing tumors [57][58][63].
ATRX is a frequent somatically mutated gene in PPGLs. The most frequent ATRX alterations have been observed in SDHx-mutated tumors, including metastasizing PPGLs [64]. Moreover, several studies showed that the somatic ATRX variant occurring with the SDHB mutation and/or TERT overexpression was an indicative marker of metastasizing tumors [65][66]. An important role of ATRX and telomere maintenance mechanisms during tumor progression was also confirmed by the presence of alternative lengthening of telomeres (ALT) in ATRX-mutated metastasizing PPGLs [67].
The Ki-67 protein is another important biomarker of tumor progression used in grading systems and prognosis prediction for several types of cancer [68]. It is also included in the pathological grading system GAPP for the estimation of metastatic potential in PPGLs. PPGLs usually have low proliferation activity with the Ki-67 score varying from 0% to 2%. However, elevated proliferation activity (over 2%) was observed in metastasizing PHEOs and PGLs [22][23][69]. Moreover, a series of studies have reported metastasizing PPGLs with the Ki-67 index of more than 4% [21][70]. Nevertheless, metastatic tumors can also have proliferation activity up to 2%, indicating the high specificity but low sensitivity of the method [71][72]. Recent research by Guo et al. showed an association between the Ki-67 index and the programmed death ligand 1 (PD-L1) expression in PPGLs [73]. Although PD-L1 expression was not significantly correlated with the presence of distant metastases, PD-L1 positivity in tumor cells with high Ki-67 may indicate that cells acquire the ability to escape the immune system, contributing to tumor growth, invasion, and metastasis [73][74]. The association of tumor progression with immune evasion in PPGLs was confirmed by the fact that almost half of the metastasizing PPGLs expressed PD-L1 or PD-L2 [74]. Additionally, the TCGA project study found a positive correlation of the Ki-67 index with metastasizing PPGLs and its highest expression in MAML3 fusion-positive tumors related to the Wnt signaling cluster. This indicates that the activation of the Wnt signaling pathway can promote tumor cell proliferation and progression of paragangliomas [75].
3. Conclusions
| Potential Marker | Characteristics Associated with Malignancy |
|---|---|
| Histopathological markers | |
| Grading system for adrenal pheochromocytoma and paraganglioma (GAPP) | Well-differentiated and moderately differentiated tumors |
| Tumor size and weight | On average, larger than 10 cm and more than 500 g |
| Adrenal gland scaled score (PASS) | ≥4 |
| Ki-67 proliferation index | >2% |
| Sustentacular cells | Cell density depletion or absent |
| Galectin-3 (LGALS3) | Increased expression detected using IHC staining |
| Succinate dehydrogenase complex subunit B (SDHB) | Negative or weak diffuse IHC staining |
| Heparanase-1 (HPSE1) | Positive IHC staining |
| Cyclooxygenase-2 (COX2) | |
| Genetic markers | |
| Succinate dehydrogenase complex subunit B (SDHB) | Germline mutation |
| Succinate dehydrogenase complex subunit D (SDHD) | |
| Fumarate hydratase (FH) | |
| Solute carrier family 25 member 11 (SLC25A11) | |
| Malate dehydrogenase 2 (MDH2) | |
| ATRX chromatin remodeler (ATRX) | Somatic mutation |
| Histone-lysine N-methyltransferase SETD2 (SETD2) | |
| Telomerase reverse transcriptase (hTERT) | |
| Mastermind-like transcriptional coactivator 3 (MAML3) | Fusion gene |
| CpG island methylator phenotype (CIMP) | High CIMP |
| MicroRNA miR-15a | Downregulation |
| Phenylethanolamine N-methyltransferase (PNMT) | |
| MicroRNA miR-483-5p | Overexpression |
| MicroRNA miR-101 | |
| MicroRNA miR-210 | |
| MicroRNA miR-21-3p | |
| MicroRNA miR-183-5p | |
| Telomerase reverse transcriptase (hTERT) | |
| Biochemical markers | |
| Normetanephrine and 3-methoxytyramine | Increased plasma or urine level |
| Neuron-specific enolase (NSE) | Increased serum level |
References
- Lloyd, R.V. Endocrine Pathology; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2010.
- Barnes, L.; Universitäts-Spital, Z.; Department, P.; World Health Organization; International Agency for Research on cancer. Pathology and Genetics of Head and Neck Tumours; WHO: Geneva, Switzerland, 2017.
- Klein, R.D.; Jin, L.; Rumilla, K.; Young, W.F., Jr.; Lloyd, R.V. Germline SDHB mutations are common in patients with apparently sporadic sympathetic paragangliomas. Diagn. Mol. Pathol. 2008, 17, 94–100.
- El-Naggar, A.K.; Chan, J.K.C.; Rubin Grandis, J.; Takata, T.; Slootweg, P.J.; International Agency for Research on Cancer. WHO Classification of Head and Neck Tumours; WHO: Geneva, Switzerland, 2017.
- Pellitteri, P.K.; Rinaldo, A.; Myssiorek, D.; Gary Jackson, C.; Bradley, P.J.; Devaney, K.O.; Shaha, A.R.; Netterville, J.L.; Manni, J.J.; Ferlito, A. Paragangliomas of the head and neck. Oral Oncol. 2004, 40, 563–575.
- van Duinen, N.; Corssmit, E.P.; de Jong, W.H.; Brookman, D.; Kema, I.P.; Romijn, J.A. Plasma levels of free metanephrines and 3-methoxytyramine indicate a higher number of biochemically active HNPGL than 24-h urinary excretion rates of catecholamines and metabolites. Eur. J. Endocrinol. 2013, 169, 377–382.
- Chapman, D.B.; Lippert, D.; Geer, C.P.; Edwards, H.D.; Russell, G.B.; Rees, C.J.; Browne, J.D. Clinical, histopathologic, and radiographic indicators of malignancy in head and neck paragangliomas. Otolaryngol. Head Neck Surg. 2010, 143, 531–537.
- Mediouni, A.; Ammari, S.; Wassef, M.; Gimenez-Roqueplo, A.P.; Laredo, J.D.; Duet, M.; Tran Ba Huy, P.; Oker, N. Malignant head/neck paragangliomas. Comparative study. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2014, 131, 159–166.
- Lloyd, R.V.; Or, K.G.; Rosai, J. WHO Classification of Tumours of Endocrine Organs; WHO: Geneva, Switzerland, 2017.
- Lee, J.H.; Barich, F.; Karnell, L.H.; Robinson, R.A.; Zhen, W.K.; Gantz, B.J.; Hoffman, H.T.; American College of Surgeons Commission on Cancer; American Cancer Society. National Cancer Data Base report on malignant paragangliomas of the head and neck. Cancer 2002, 94, 730–737.
- Rodriguez-Cuevas, S.; Lopez-Garza, J.; Labastida-Almendaro, S. Carotid body tumors in inhabitants of altitudes higher than 2000 meters above sea level. Head Neck 1998, 20, 374–378.
- Williams, M.D. Paragangliomas of the Head and Neck: An Overview from Diagnosis to Genetics. Head Neck Pathol. 2017, 11, 278–287.
- Magliulo, G.; Zardo, F.; Varacalli, S.; D’Amico, R. Multiple paragangliomas of the head and neck. Otorrinolaringol. Ibero Am. 2003, 30, 31–38.
- Zanoletti, E.; Mazzoni, A. Vagal paraganglioma. Skull Base 2006, 16, 161–167.
- Hamidi, O.; Young, W.F., Jr.; Iniguez-Ariza, N.M.; Kittah, N.E.; Gruber, L.; Bancos, C.; Tamhane, S.; Bancos, I. Malignant Pheochromocytoma and Paraganglioma: 272 Patients Over 55 Years. J. Clin. Endocrinol. Metab. 2017, 102, 3296–3305.
- Li, M.; Pamporaki, C.; Fliedner, S.M.J.; Timmers, H.J.L.M.; Nölting, S.; Beuschlein, F.; Prejbisz, A.; Remde, H.; Robledo, M.; Bornstein, S.R.; et al. Metastatic pheochromocytoma and paraganglioma: Signs and symptoms related to catecholamine secretion. Discov. Oncol. 2021, 12, 9.
- Kiernan, C.M.; Solorzano, C.C. Pheochromocytoma and Paraganglioma: Diagnosis, Genetics, and Treatment. Surg. Oncol. Clin. N. Am. 2016, 25, 119–138.
- Abdel-Aziz, T.; Chung, T.-T.; Bomanji, J.; Gaze, M.; Kurzawinski, T. Patterns of recurrence, response to treatment and mortality in patients with malignant phaeochromocytomas and paragangliomas—A single centre experience. Endocr. Abstr. 2017.
- Contrera, K.J.; Yong, V.; Reddy, C.A.; Liu, S.W.; Lorenz, R.R. Recurrence and Progression of Head and Neck Paragangliomas after Treatment. Otolaryngol. Head Neck Surg. 2020, 162, 504–511.
- Khadilkar, K.; Sarathi, V.; Kasaliwal, R.; Pandit, R.; Goroshi, M.; Malhotra, G.; Dalvi, A.; Bakshi, G.; Bhansali, A.; Rajput, R.; et al. Predictors of malignancy in patients with pheochromocytomas/paragangliomas: Asian Indian experience. Endocr. Connect 2016, 5, 89–97.
- de Wailly, P.; Oragano, L.; Rade, F.; Beaulieu, A.; Arnault, V.; Levillain, P.; Kraimps, J.L. Malignant pheochromocytoma: New malignancy criteria. Langenbecks Arch Surg. 2012, 397, 239–246.
- Ohji, H.; Sasagawa, I.; Iciyanagi, O.; Suzuki, Y.; Nakada, T. Tumour angiogenesis and Ki-67 expression in phaeochromocytoma. BJU Int. 2001, 87, 381–385.
- van der Harst, E.; Bruining, H.A.; Jaap Bonjer, H.; van der Ham, F.; Dinjens, W.N.; Lamberts, S.W.; de Herder, W.W.; Koper, J.W.; Stijnen, T.; Proye, C.; et al. Proliferative index in phaeochromocytomas: Does it predict the occurrence of metastases? J. Pathol. 2000, 191, 175–180.
- Thompson, L.D. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: A clinicopathologic and immunophenotypic study of 100 cases. Am. J. Surg. Pathol. 2002, 26, 551–566.
- Kim, K.Y.; Kim, J.H.; Hong, A.R.; Seong, M.W.; Lee, K.E.; Kim, S.J.; Kim, S.W.; Shin, C.S.; Kim, S.Y. Disentangling of Malignancy from Benign Pheochromocytomas/Paragangliomas. PLoS ONE 2016, 11, e0168413.
- Lenders, J.W.; Duh, Q.Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.P.; Grebe, S.K.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F., Jr.; Endocrine, S. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942.
- Plouin, P.F.; Amar, L.; Dekkers, O.M.; Fassnacht, M.; Gimenez-Roqueplo, A.P.; Lenders, J.W.; Lussey-Lepoutre, C.; Steichen, O.; Guideline Working, G. European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur. J. Endocrinol. 2016, 174, G1–G10.
- Feng, F.; Zhu, Y.; Wang, X.; Wu, Y.; Zhou, W.; Jin, X.; Zhang, R.; Sun, F.; Kasoma, Z.; Shen, Z. Predictive factors for malignant pheochromocytoma: Analysis of 136 patients. J. Urol. 2011, 185, 1583–1590.
- Amar, L.; Peyrard, S.; Rossignol, P.; Zinzindohoue, F.; Gimenez-Roqueplo, A.P.; Plouin, P.F. Changes in urinary total metanephrine excretion in recurrent and malignant pheochromocytomas and secreting paragangliomas. Ann. N. Y. Acad. Sci. 2006, 1073, 383–391.
- Gupta, P.; Khurana, M.L.; Khadgawat, R.; Bal, C.S.; Kumar, G.; Sharma, S.C.; Tandon, N. Plasma free metanephrine, normetanephrine, and 3-methoxytyramine for the diagnosis of pheochromocytoma/paraganglioma. Indian J. Endocrinol. Metab. 2015, 19, 633–638.
- Eisenhofer, G.; Lenders, J.W.; Siegert, G.; Bornstein, S.R.; Friberg, P.; Milosevic, D.; Mannelli, M.; Linehan, W.M.; Adams, K.; Timmers, H.J.; et al. Plasma methoxytyramine: A novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur. J. Cancer 2012, 48, 1739–1749.
- Timmers, H.J.; Kozupa, A.; Eisenhofer, G.; Raygada, M.; Adams, K.T.; Solis, D.; Lenders, J.W.; Pacak, K. Clinical presentations, biochemical phenotypes, and genotype-phenotype correlations in patients with succinate dehydrogenase subunit B-associated pheochromocytomas and paragangliomas. J. Clin. Endocrinol. Metab. 2007, 92, 779–786.
- Cavadas, C.; Silva, A.P.; Mosimann, F.; Cotrim, M.D.; Ribeiro, C.A.; Brunner, H.R.; Grouzmann, E. NPY regulates catecholamine secretion from human adrenal chromaffin cells. J. Clin. Endocrinol. Metab. 2001, 86, 5956–5963.
- DeS, Senanayake, P.; Denker, J.; Bravo, E.L.; Graham, R.M. Production, characterization, and expression of neuropeptide Y by human pheochromocytoma. J. Clin. Investig. 1995, 96, 2503–2509.
- Grouzmann, E.; Comoy, E.; Bohuon, C. Plasma neuropeptide Y concentrations in patients with neuroendocrine tumors. J. Clin. Endocrinol. Metab. 1989, 68, 808–813.
- Grouzmann, E.; Gicquel, C.; Plouin, P.F.; Schlumberger, M.; Comoy, E.; Bohuon, C. Neuropeptide Y and neuron-specific enolase levels in benign and malignant pheochromocytomas. Cancer 1990, 66, 1833–1835.
- Kuvshinoff, B.W.; Nussbaum, M.S.; Richards, A.I.; Bloustein, P.; McFadden, D.W. Neuropeptide Y secretion from a malignant extraadrenal retroperitoneal paraganglioma. Cancer 1992, 70, 2350–2353.
- Helman, L.J.; Cohen, P.S.; Averbuch, S.D.; Cooper, M.J.; Keiser, H.R.; Israel, M.A. Neuropeptide Y expression distinguishes malignant from benign pheochromocytoma. J. Clin. Oncol. 1989, 7, 1720–1725.
- Thouennon, E.; Elkahloun, A.G.; Guillemot, J.; Gimenez-Roqueplo, A.P.; Bertherat, J.; Pierre, A.; Ghzili, H.; Grumolato, L.; Muresan, M.; Klein, M.; et al. Identification of potential gene markers and insights into the pathophysiology of pheochromocytoma malignancy. J. Clin. Endocrinol. Metab. 2007, 92, 4865–4872.
- Korner, M.; Waser, B.; Reubi, J.C. High expression of neuropeptide y receptors in tumors of the human adrenal gland and extra-adrenal paraganglia. Clin. Cancer Res. 2004, 10, 8426–8433.
- Oishi, S.; Sato, T. Elevated serum neuron-specific enolase in patients with malignant pheochromocytoma. Cancer 1988, 61, 1167–1170.
- Tischler, A.S.; deKrijger, R.R. 15 YEARS OF PARAGANGLIOMA: Pathology of pheochromocytoma and paraganglioma. Endocr. Relat. Cancer 2015, 22, T123–T133.
- Marotta, V.; Zatelli, M.C.; Sciammarella, C.; Ambrosio, M.R.; Bondanelli, M.; Colao, A.; Faggiano, A. Chromogranin A as circulating marker for diagnosis and management of neuroendocrine neoplasms: More flaws than fame. Endocr. Relat. Cancer 2018, 25, R11–R29.
- Bilek, R.; Vlcek, P.; Safarik, L.; Michalsky, D.; Novak, K.; Duskova, J.; Vaclavikova, E.; Widimsky, J., Jr.; Zelinka, T. Chromogranin A in the Laboratory Diagnosis of Pheochromocytoma and Paraganglioma. Cancers 2019, 11, 586.
- Grossrubatscher, E.; Dalino, P.; Vignati, F.; Gambacorta, M.; Pugliese, R.; Boniardi, M.; Rossetti, O.; Marocchi, A.; Bertuzzi, M.; Loli, P. The role of chromogranin A in the management of patients with phaeochromocytoma. Clin. Endocrinol. 2006, 65, 287–293.
- Kimura, N.; Miura, W.; Noshiro, T.; Mizunashi, K.; Hanew, K.; Shimizu, K.; Watanabe, T.; Shibukawa, S.; Sohn, H.E.; Abe, K.; et al. Plasma chromogranin A in pheochromocytoma, primary hyperparathyroidism and pituitary adenoma in comparison with catecholamine, parathyroid hormone and pituitary hormones. Endocr. J. 1997, 44, 319–327.
- Szalat, A.; Fraenkel, M.; Doviner, V.; Salmon, A.; Gross, D.J. Malignant pheochromocytoma: Predictive factors of malignancy and clinical course in 16 patients at a single tertiary medical center. Endocrine 2011, 39, 160–166.
- Parisien-La Salle, S.; Provencal, M.; Bourdeau, I. Chromogranin A in a Cohort of Pheochromocytomas and Paragangliomas: Usefulness at Diagnosis and as an Early Biomarker of Recurrence. Endocr. Pract. 2021, 27, 318–325.
- Hescot, S.; Curras-Freixes, M.; Deutschbein, T.; van Berkel, A.; Vezzosi, D.; Amar, L.; de la Fouchardiere, C.; Valdes, N.; Riccardi, F.; Do Cao, C.; et al. Prognosis of Malignant Pheochromocytoma and Paraganglioma (MAPP-Prono Study): A European Network for the Study of Adrenal Tumors Retrospective Study. J. Clin. Endocrinol. Metab. 2019, 104, 2367–2374.
- McCrary, H.C.; Babajanian, E.; Calquin, M.; Carpenter, P.; Casazza, G.; Naumer, A.; Greenberg, S.; Kohlmann, W.; Cannon, R.; Monroe, M.M.; et al. Characterization of Malignant Head and Neck Paragangliomas at a Single Institution Across Multiple Decades. JAMA Otolaryngol. Head Neck Surg. 2019, 145, 641–646.
- van Hulsteijn, L.T.; Dekkers, O.M.; Hes, F.J.; Smit, J.W.; Corssmit, E.P. Risk of malignant paraganglioma in SDHB-mutation and SDHD-mutation carriers: A systematic review and meta-analysis. J. Med. Genet. 2012, 49, 768–776.
- Neumann, H.P.; Pawlu, C.; Peczkowska, M.; Bausch, B.; McWhinney, S.R.; Muresan, M.; Buchta, M.; Franke, G.; Klisch, J.; Bley, T.A.; et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 2004, 292, 943–951.
- Benn, D.E.; Gimenez-Roqueplo, A.P.; Reilly, J.R.; Bertherat, J.; Burgess, J.; Byth, K.; Croxson, M.; Dahia, P.L.; Elston, M.; Gimm, O.; et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J. Clin. Endocrinol. Metab. 2006, 91, 827–836.
- Snezhkina, A.V.; Fedorova, M.S.; Pavlov, V.S.; Kalinin, D.V.; Golovyuk, A.L.; Pudova, E.A.; Guvatova, Z.G.; Melnikova, N.V.; Dmitriev, A.A.; Razmakhaev, G.S.; et al. Mutation Frequency in Main Susceptibility Genes Among Patients with Head and Neck Paragangliomas. Front. Genet. 2020, 11, 614908.
- Burnichon, N.; Rohmer, V.; Amar, L.; Herman, P.; Leboulleux, S.; Darrouzet, V.; Niccoli, P.; Gaillard, D.; Chabrier, G.; Chabolle, F.; et al. The succinate dehydrogenase genetic testing in a large prospective series of patients with paragangliomas. J. Clin. Endocrinol. Metab. 2009, 94, 2817–2827.
- Hensen, E.F.; Siemers, M.D.; Jansen, J.C.; Corssmit, E.P.; Romijn, J.A.; Tops, C.M.; van der Mey, A.G.; Devilee, P.; Cornelisse, C.J.; Bayley, J.P.; et al. Mutations in SDHD are the major determinants of the clinical characteristics of Dutch head and neck paraganglioma patients. Clin. Endocrinol. 2011, 75, 650–655.
- Castro-Vega, L.J.; Buffet, A.; De Cubas, A.A.; Cascon, A.; Menara, M.; Khalifa, E.; Amar, L.; Azriel, S.; Bourdeau, I.; Chabre, O.; et al. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum. Mol. Genet. 2014, 23, 2440–2446.
- Buffet, A.; Morin, A.; Castro-Vega, L.J.; Habarou, F.; Lussey-Lepoutre, C.; Letouze, E.; Lefebvre, H.; Guilhem, I.; Haissaguerre, M.; Raingeard, I.; et al. Germline Mutations in the Mitochondrial 2-Oxoglutarate/Malate Carrier SLC25A11 Gene Confer a Predisposition to Metastatic Paragangliomas. Cancer Res. 2018, 78, 1914–1922.
- Cascon, A.; Comino-Mendez, I.; Curras-Freixes, M.; de Cubas, A.A.; Contreras, L.; Richter, S.; Peitzsch, M.; Mancikova, V.; Inglada-Perez, L.; Perez-Barrios, A.; et al. Whole-exome sequencing identifies MDH2 as a new familial paraganglioma gene. J. Natl. Cancer Inst. 2015, 107.
- Alrezk, R.; Suarez, A.; Tena, I.; Pacak, K. Update of Pheochromocytoma Syndromes: Genetics, Biochemical Evaluation, and Imaging. Front. Endocrinol. 2018, 9, 515.
- Letouze, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 2013, 23, 739–752.
- Castro-Vega, L.J.; Letouze, E.; Burnichon, N.; Buffet, A.; Disderot, P.H.; Khalifa, E.; Loriot, C.; Elarouci, N.; Morin, A.; Menara, M.; et al. Multi-omics analysis defines core genomic alterations in pheochromocytomas and paragangliomas. Nat. Commun. 2015, 6, 6044.
- Calsina, B.; Curras-Freixes, M.; Buffet, A.; Pons, T.; Contreras, L.; Leton, R.; Comino-Mendez, I.; Remacha, L.; Calatayud, M.; Obispo, B.; et al. Role of MDH2 pathogenic variant in pheochromocytoma and paraganglioma patients. Genet. Med. 2018, 20, 1652–1662.
- Fishbein, L.; Khare, S.; Wubbenhorst, B.; DeSloover, D.; D’Andrea, K.; Merrill, S.; Cho, N.W.; Greenberg, R.A.; Else, T.; Montone, K.; et al. Whole-exome sequencing identifies somatic ATRX mutations in pheochromocytomas and paragangliomas. Nat. Commun. 2015, 6, 6140.
- Job, S.; Draskovic, I.; Burnichon, N.; Buffet, A.; Cros, J.; Lepine, C.; Venisse, A.; Robidel, E.; Verkarre, V.; Meatchi, T.; et al. Telomerase Activation and ATRX Mutations Are Independent Risk Factors for Metastatic Pheochromocytoma and Paraganglioma. Clin. Cancer Res. 2019, 25, 760–770.
- Irwin, T.; Konnick, E.Q.; Tretiakova, M.S. Malignant Intrarenal/Renal Pelvis Paraganglioma with Co-Occurring SDHB and ATRX Mutations. Endocr. Pathol. 2019, 30, 270–275.
- Comino-Mendez, I.; Tejera, A.M.; Curras-Freixes, M.; Remacha, L.; Gonzalvo, P.; Tonda, R.; Leton, R.; Blasco, M.A.; Robledo, M.; Cascon, A. ATRX driver mutation in a composite malignant pheochromocytoma. Cancer Genet. 2016, 209, 272–277.
- Sun, X.; Kaufman, P.D. Ki-67: More than a proliferation marker. Chromosoma 2018, 127, 175–186.
- Chang, J.; Kim, S.-H.; Min, H.S.; Kim, H.-Y.; Jung, S.-E.; Park, K.-W.; Lee, S.-C. Risk Factors for Malignancy of Pheochromocytoma and Abdominal Paraganglioma in Children: Clinicopathologic Perspectives. J. Korean Assoc. Pediatr. Surg. 2013, 19, 108–121.
- Clarke, M.R.; Weyant, R.J.; Watson, C.G.; Carty, S.E. Prognostic markers in pheochromocytoma. Hum. Pathol. 1998, 29, 522–526.
- Strong, V.E.; Kennedy, T.; Al-Ahmadie, H.; Tang, L.; Coleman, J.; Fong, Y.; Brennan, M.; Ghossein, R.A. Prognostic indicators of malignancy in adrenal pheochromocytomas: Clinical, histopathologic, and cell cycle/apoptosis gene expression analysis. Surgery 2008, 143, 759–768.
- Kovacs, K.; Bell, D.; Gardiner, G.W.; Honey, R.J.; Goguen, J.; Rotondo, F. Malignant paraganglioma of the urinary bladder: Immunohistochemical study of prognostic indicators. Endocr. Pathol. 2005, 16, 363–369.
- Guo, D.; Zhao, X.; Wang, A.; Xie, Q.; Xu, X.; Sun, J. PD-L1 expression and association with malignant behavior in pheochromocytomas/paragangliomas. Hum. Pathol. 2019, 86, 155–162.
- Pinato, D.J.; Black, J.R.; Trousil, S.; Dina, R.E.; Trivedi, P.; Mauri, F.A.; Sharma, R. Programmed cell death ligands expression in phaeochromocytomas and paragangliomas: Relationship with the hypoxic response, immune evasion and malignant behavior. Oncoimmunology 2017, 6, e1358332.
- Fishbein, L.; Leshchiner, I.; Walter, V.; Danilova, L.; Robertson, A.G.; Johnson, A.R.; Lichtenberg, T.M.; Murray, B.A.; Ghayee, H.K.; Else, T.; et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell 2017, 31, 181–193.

