 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lidia Ciccone | + 2580 word(s) | 2580 | 2021-10-26 10:40:41 | | | |
| 2 | Beatrix Zheng | Meta information modification | 2580 | 2021-11-02 11:10:56 | | |
Video Upload Options
Carbonic anhydrases (CAs) are a group of ubiquitously expressed metalloenzymes that catalyze the reversible hydration/dehydration of CO2/HCO3. Thus, they are involved in those physiological and pathological processes in which cellular pH buffering plays a relevant role. The inhibition of CAs has pharmacologic applications for several diseases.
1. Introduction
According to the International League Against Epilepsy (ILAE), epilepsy is a chronic brain disorder operationally defined by the occurrence of two unprovoked seizures more than 24 h apart, or one unprovoked seizure when the risk for another is known to be high (>60%) [1]. Seizures can manifest in a variety of different clinical presentations with motor, sensory, autonomic or psychic origin [2]. Seizure episodes are a result of abnormal excessive or synchronous neural activity in the brain. Seizures are classified into focal and generalized types. Focal seizures are localized in a specific cerebral area. Thus, the behavioral outcome depends on the brain regions where synchronous firing of a neuronal cell group occurs. Generalized seizures, spreading through thalamocortical connections, involve both cerebral hemispheres. Considering the specific symptoms and etiology, subclassifications of epileptic seizures are also reported [3].
The high impact of the disease on global health has provoked immense efforts from the scientific community to shed light on the complex mechanisms underlying seizure generation and to develop therapeutic strategies to pharmacologically treat epilepsy. However, antiepileptic drugs (AED) currently available and employed in clinical practice can treat only some subtypes of epilepsy and, often, pharmacological treatment may not be resolute [4]. For this reason, there is an urgent need to identify new molecular targets in order to expand the therapeutic options to treat and to defeat this dramatic pathology [5][6][7].
2. CAs and Their Role in Epilepsy
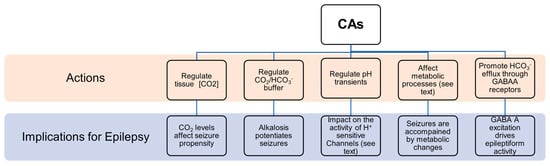
- (1)
-
Seizures are accompanied by pronounced changes in ionic composition in brain compartments and by pH shift that, directly or indirectly, influence the concentration of the chemical species of the reaction catalyzed by CAs.
- (2)
-
CAs regulate CO2 tissue concentration, and it has been demonstrated that CO2 has a role in epilepsy. In particular, clinical evidence suggest that the enhancement of CO2 concentration results in better seizure control [21], while low CO2 levels are linked to higher seizure propensity [22]. The inhibition of CAs resulted in increased CO2 concentration and a positive outcome in epilepsy management [23].
- (3)
-
It has been clearly shown that alkalosis generally potentiates seizures by increasing neuronal excitability, while acidosis has an opposite effect [24]. Since their role is in the regulation of the CO2/ HCO3− buffer system, CAs are crucially involved in the control of neural excitability [25]. For instance, it has been demonstrated that CA IV and CA XIV play a role in extracellular buffering in response to neural activity [26].
- (4)
-
Mitochondrial dysfunction has been identified as one potential cause of epileptic seizures [27]. There is a vicious cycle between mitochondrial dysfunction and epileptic seizures because seizures themselves can induce mitochondrial damage that consequently triggers seizures [27]. It is known that CAs are involved in mitochondria biogenesis and physiology, and in glucose and lipid metabolism in human Sertori cells [28]. In particular, CA V A and CA V B are specifically localized in mitochondria. They hydrate carbon dioxide to yield bicarbonate ions and a proton that contribute to normal mitochondria metabolism. In the nervous system, CA V is expressed in astrocytes as well as in neurons. It has been proposed that CA V in neurons could be involved in the regulation of the intra-mitochondrial Ca2+ levels, thus contributing to the stability of the intracellular calcium concentration preventing neuronal degeneration and cell death [13]. Another possible function of CA V is to participate in the regulation of neuronal HCO3− homeostasis taking part in physiological neuronal function. Moreover, it has been reported that the intracellular regeneration of HCO3− and its elimination from the extracellular environment results in a repolarization in GABA responses, suggesting that CA V might also be involved in neuronal transmission [13][29].
- (5)
-
Regulating the kinetics of pH transients [25][30][31]. CAs can influence the function of a broad array of proton-sensitive transmembrane proteins implicated in neuronal signaling such as GABAARs [30][31], N-methyl-D-aspartate (NMDA) receptors [32][33], H+-gated channels [34] and cation channels [35][36]. For example, the activity of excitatory receptors for glutamate, NMDA receptors, is inhibited by extracellular protons [37]. The initial seizure-associated extracellular alkaline shift, apparently influenced by CA activity [26], likely sustains NMDA receptors’ activation during seizures. Moreover, it has been shown that CA XIV, located in close vicinity to the NMDA receptor at the synapses, regulates pH transients in the perisynaptic microenvironment and their impact on NMDA receptors’ activity [33].
- (6)
-
It has been shown that glycolysis increases during seizures and that the glycolytic metabolite lactic acid can be used as an energy source [38]. A specific isoform of CAs facilitate lactate transport in astrocytes as well as in neurons [39]. In addition, CAs can intervene in lactic acid-induced acidosis, that seems to be implicated in seizure termination [38][40]. Moreover, CAs provide substrates required for the function of metabolic enzymes involved in epilepsy. For instance, a failure in pyruvate carboxylase (PC) function may lead to seizures, as demonstrated by the fact that PC deficiency is related to recurrent seizures in patients. CA V, providing HCO3− to pyruvate carboxylase, is involved in controlling the proper functioning of this enzyme [9] and, then, its action might have implications for epilepsy.
- (7)
-
Numerous experimental and clinical studies support the notion that oxidative stress substantially contributes to the pathogenesis of epilepsy [41]. Studies showed that patients affected by epilepsy report a remarkable increase in levels of oxidative markers, such as malondialdehyde (MDA), protein carbonylation (PC) and nitric oxide (NO), when compared to a control group. An excessive production of free radicals could be implicated in neuronal hyperexcitability that triggers epileptogenesis. Moreover, it has been reported that overproduction of reactive oxygen species (ROS) provokes the progressive disruption of Ca2+ homeostasis essential for neuronal survival. In this context, it has been proposed that CAs, in particular CA VII might also have a role in the cell defence against oxidative damage thanks to its cysteine residues [42].
- (8)
-
GABAergic inhibition has been traditionally considered as the principal mechanism counterbalancing glutamatergic excitation and preventing epileptiform activity. Indeed, many of the currently used antiepileptic drugs act through enhancement of GABAergic signaling. However, much evidence has shown that epileptiform events can also be characterized by synchronous firing driven by excitatory GABA [43]. As during the first phases of development [44], excitatory action of GABA in epilepsy is due to (a) elevated intracellular chloride concentration as a result of chloride accumulation during hyperactivity [45]. High levels of intra-neuronal Cl− leads to Cl− efflux and then to depolarization in response to GABA binding to its type A receptor; (b) HCO3− permeability of GABA-A channels [46][47] that causes HCO3− efflux and then depolarization; (c) elevation of extracellular potassium caused by KCC2-mediated extrusion of chloride and potassium that results in membrane depolarization [48]. CAs are implicated in this abnormal epilepsy-associated GABA-A excitation. Specifically, it has been shown that they have a role in favouring the efflux of HCO3− ions through GABA-A receptors [49][50]. CA VII, which plays an important role in the development of febrile seizures [20], has been identified as a key molecule in GABAergic excitation and it has been suggested that CA VII developmental expression governs the electrophysiological behaviour related to neural circuit plasticity and to susceptibility to epileptogenesis [51].
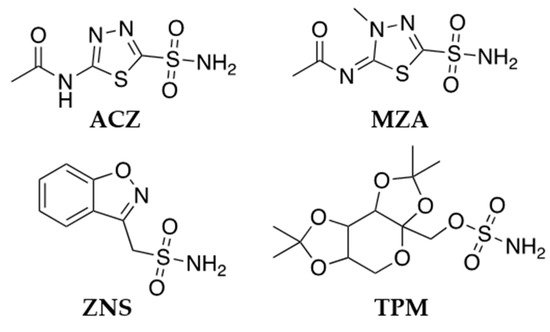
3. CA Inhibitors Clinically Employed in Epilepsy Therapy
References
- Definition of Epilepsy 2014. International League Against Epilepsy. Available online: https://www.ilae.org/guidelines/definition-and-classification/definition-of-epilepsy-2014 (accessed on 30 June 2021).
- Shakirullah, A.N.; Khan, A.; Nabi, M. The Prevalence, Incidence and Etiology of Epilepsy. Int. J. Clin. Exp. Neurol. 2014, 2, 29–39.
- Sarmast, S.T.; Abdullahi, A.M.; Jahan, N. Current Classification of Seizures and Epilepsies: Scope, Limitations and Recommendations for Future Action. Cureus 2020, 12, e10549.
- Löscher, W.; Potschka, H.; Sisodiya, S.M.; Vezzani, A. Drug Resistance in Epilepsy: Clinical Impact, Potential Mechanisms, and New Innovative Treatment Options. Pharmacol. Rev. 2020, 72, 606–638.
- Epilepsy. Available online: https://www.who.int/news-room/fact-sheets/detail/epilepsy (accessed on 19 July 2021).
- Cerri, C.; Genovesi, S.; Allegra, M.; Pistillo, F.; Püntener, U.; Guglielmotti, A.; Perry, V.H.; Bozzi, Y.; Caleo, M. The Chemokine CCL2 Mediates the Seizure-enhancing Effects of Systemic Inflammation. J. Neurosci. 2016, 36, 3777–3788.
- Cerri, C.; Caleo, M.; Bozzi, Y. Chemokines as New Inflammatory Players in the Pathogenesis of Epilepsy. Epilepsy Res. 2017, 136, 77–83.
- Pastorekova, S.; Parkkila, S.; Pastorek, J.; Supuran, C.T. Review Article. J. Enzym. Inhib. Med. Chem. 2004, 19, 199–229.
- Anne, T.; Jean-Michel, D.; Claudiu, T.S.; Bernard, M. Carbonic Anhydrase Inhibitors as Anticonvulsant Agents. Curr. Top. Med. Chem. 2007, 7, 855–864.
- Ozsoy, H.Z. Anticonvulsant Effects of Carbonic Anhydrase Inhibitors: The Enigmatic Link Between Carbonic Anhydrases and Electrical Activity of the Brain. Neurochem. Res. 2021, 46, 2783–2799.
- Kida, E.; Palminiello, S.; Golabek, A.A.; Walus, M.; Wierzba-Bobrowicz, T.; Rabe, A.; Albertini, G.; Wisniewski, K.E. Carbonic Anhydrase II in the Developing and Adult Human Brain. J. Neuropathol. Exp. Neurol. 2006, 65, 664–674.
- Mishra, C.B.; Kumari, S.; Angeli, A.; Bua, S.; Tiwari, M.; Supuran, C.T. Discovery of Benzenesulfonamide Derivatives as Carbonic Anhydrase Inhibitors with Effective Anticonvulsant Action: Design, Synthesis, and Pharmacological Evaluation. J. Med. Chem. 2018, 61, 3151–3165.
- Ghandour, M.S.; Parkkila, A.-K.; Parkkila, S.; Waheed, A.; Sly, W.S. Mitochondrial Carbonic Anhydrase in the Nervous System. J. Neurochem. 2000, 75, 2212–2220.
- Taniuchi, K.; Nishimori, I.; Takeuchi, T.; Fujikawa-Adachi, K.; Ohtsuki, Y.; Onishi, S. Developmental Expression of Carbonic Anhydrase-Related Proteins VIII, X, And XI in the Human Brain. Neuroscience 2002, 112, 93–99.
- Harju, A.-K.; Bootorabi, F.; Kuuslahti, M.; Supuran, C.T.; Parkkila, S. Carbonic anhydrase III: A Neglected Isozyme is Stepping into the Limelight. J. Enzym. Inhib. Med. Chem. 2013, 28, 231–239.
- Aspatwar, A.; Tolvanen, M.E.E.; Ortutay, C.; Parkkila, S. Carbonic Anhydrase Related Proteins: Molecular Biology and Evolution. Carbon. Anhydrase 2014, 75, 135–156.
- Hilvo, M.; Tolvanen, M.; Clark, A.; Shen, B.; Shah, G.N.; Waheed, A.; Halmi, P.; Hänninen, M.; Hämäläinen, J.M.; Vihinen, M.; et al. Characterization of CA XV, a new GPI-anchored form of Carbonic Anhydrase. Biochem. J. 2005, 392, 83–92.
- Parkkila, S.; Parkkila, A.-K.; Rajaniemi, H.; Shah, G.N.; Grubb, J.H.; Waheed, A.; Sly, W.S. Expression of Membrane-Associated Carbonic Anhydrase XIV on Neurons and Axons in Mouse and Human Brain. Proc. Natl. Acad. Sci. USA 2001, 98, 1918–1923.
- Halmi, P.; Parkkila, S.; Honkaniemi, J. Expression of Carbonic Anhydrases II, IV, VII, VIII and XII in Rat Brain After Kainic Acid Induced Status Epilepticus. Neurochem. Int. 2006, 48, 24–30.
- Ruusuvuori, E.; Huebner, A.K.; Kirilkin, I.; Yukin, A.Y.; Blaesse, P.; Helmy, M.; Kang, H.J.; El Muayed, M.; Hennings, J.C.; Voipio, J.; et al. Neuronal Carbonic Anhydrase VII Provides Gabaergic Excitatory Drive to Exacerbate Febrile Seizures. EMBO J. 2013, 32, 2275–2286.
- Miller, J.W. Stopping Seizures with Carbon Dioxide. Epilepsy Curr. 2011, 11, 114–115.
- Jonas, J.; Vignal, J.-P.; Baumann, C.; Anxionnat, J.-F.; Muresan, M.; Vespignani, H.; Maillard, L. Effect of Hyperventilation on Seizure Activation: Potentiation by Antiepileptic Drug Tapering. J. Neurol. Neurosurg. Psychiatry 2011, 82, 928–930.
- Aggarwal, M.; Kondeti, B.; McKenna, R. Anticonvulsant/Antiepileptic Carbonic Anhydrase Inhibitors: A Patent Review. Expert Opin. Ther. Patents 2013, 23, 717–724.
- Raimondo, J.; Burman, R.J.; Katz, A.A.; Akerman, C.J. Ion Dynamics During Seizures. Front. Cell. Neurosci. 2015, 9, 419.
- Ruusuvuori, E.; Kaila, K. Carbonic Anhydrases and Brain pH in the Control of Neuronal Excitability. In Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications; Frost, S.C., McKenna, R., Eds.; Subcellular Biochemistry; Springer: Dordrecht, The Netherlands, 2014; pp. 271–290. ISBN 978-94-007-7359-2.
- Shah, G.N.; Ulmasov, B.; Waheed, A.; Becker, T.; Makani, S.; Svichar, N.; Chesler, M.; Sly, W.S. Carbonic Anhydrase IV and XIV Knockout Mice: Roles of the Respective Carbonic Anhydrases in Buffering the Extracellular Space in Brain. Proc. Natl. Acad. Sci. USA 2005, 102, 16771–16776.
- Folbergrová, J.; Kunz, W.S. Mitochondrial dysfunction in epilepsy. Mitochondrion 2012, 12, 35–40.
- Bernardino, R.; Dias, T.R.; Moreira, B.; Cunha, M.; Barros, A.; Oliveira, E.; Sousa, M.; Alves, M.G.; Oliveira, P.F. Carbonic Anhydrases are Involved in Mitochondrial Biogenesis and Control the Production of Lactate by Human Sertoli Cells. FEBS J. 2019, 286, 1393–1406.
- Lambert, N.; Grover, L. The Mechanism of Biphasic GABA Responses. Science 1995, 269, 928–929.
- Chesler, M. Regulation and Modulation of pH in the Brain. Physiol. Rev. 2003, 83, 1183–1221.
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and Regulators of Intracellular pH. Nat. Rev. Mol. Cell Biol. 2010, 11, 50–61.
- Pasternack, M.; Smirnov, S.; Kaila, K. Proton Modulation of Functionally Distinct GABAA Receptors in Acutely Isolated Pyramidal Neurons of Rat Hippocampus. Neuropharmacology 1996, 35, 1279–1288.
- Wilkins, M.E.; Hosie, A.M.; Smart, T.G. Proton Modulation of Recombinant GABAAreceptors: Influence of GABA Concentration and the β Subunit TM2-TM3 Domain. J. Physiol. 2005, 567, 365–377.
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.; Vance, K.M.; Ogden, K.; Hansen, K.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol. Rev. 2010, 62, 405–496.
- Makani, S.; Chen, H.-Y.; Esquenazi, S.; Shah, G.N.; Waheed, A.; Sly, W.S.; Chesler, M. NMDA Receptor-Dependent Afterdepolarizations Are Curtailed by Carbonic Anhydrase 14: Regulation of a Short-Term Postsynaptic Potentiation. J. Neurosci. 2012, 32, 16754–16762.
- Waldmann, R.; Champigny, G.; Bassilana, F.; Heurteaux, C.; Lazdunski, M. A Proton-Gated Cation Channel Involved in Acid-Sensing. Nature 1997, 386, 173–177.
- Munsch, T.; Pape, H. Modulation of the Hyperpolarization-Activated Cation Current of Rat Thalamic Relay Neurones by Intracellular pH. J. Physiol. 1999, 519, 493–504.
- Enyedi, P.; Czirják, G. Molecular Background of Leak K+ Currents: Two-Pore Domain Potassium Channels. Physiol. Rev. 2010, 90, 559–605.
- Tang, C.M.; Dichter, M.; Morad, M. Modulation of the N-methyl-D-aspartate Channel by Extracellular H+. Proc. Natl. Acad. Sci. USA 1990, 87, 6445–6449.
- Yang, H.; Wu, J.; Guo, R.; Peng, Y.; Zheng, W.; Liu, D.; Song, Z. Glycolysis in Energy Metabolism During Seizures. Neural Regen. Res. 2013, 8, 1316–1326.
- Svichar, N.; Chesler, M. Surface Carbonic Anhydrase Activity on Astrocytes and Neurons Facilitates Lactate Transport. Glia 2003, 41, 415–419.
- Deitmer, J.W.; Theparambil, S.M.; Ruminot, I.; Noor, S.I.; Becker, H.M. Energy Dynamics in the Brain: Contributions of Astrocytes to Metabolism and pH Homeostasis. Front. Neurosci. 2019, 13, 1301.
- Geronzi, U.; Lotti, F.; Grosso, S. Oxidative Stress in Epilepsy. Expert Rev. Neurother. 2018, 18, 427–434.
- Ben-Ari, Y.; Gaiarsa, J.-L.; Tyzio, R.; Khazipov, R. GABA: A Pioneer Transmitter That Excites Immature Neurons and Generates Primitive Oscillations. Physiol. Rev. 2007, 87, 1215–1284.
- Monti, S.M.; Supuran, C.T.; De Simone, G.; Di Fiore, A. Chapter 9—Carbonic Anhydrase VII. In Carbonic Anhydrases as Biocatalysts; Supuran, C.T., De Simone, G., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 151–168. ISBN 978-0-444-63258-6.
- Khazipov, R. GABAergic Synchronization in Epilepsy. Cold Spring Harb. Perspect. Med. 2016, 6, a022764.
- Fujiwara-Tsukamoto, Y.; Isomura, Y.; Nambu, A.; Takada, M. Excitatory Gaba Input Directly Drives Seizure-Like Rhythmic Synhronization in Mature Hippocampal CA1 Pyramidal Cells. Neuroscience 2003, 119, 265–275.
- Kaila, K.; Lamsa, K.; Smirnov, S.; Taira, T.; Voipio, J. Long-Lasting GABA-Mediated Depolarization Evoked by High-Frequency Stimulation in Pyramidal Neurons of Rat Hippocampal Slice is Attributable to a Network-Driven, Bicarbonate-Dependent K+Transient. J. Neurosci. 1997, 17, 7662–7672.
- Staley, K.J.; Soldo, B.L.; Proctor, W.R. Ionic Mechanisms of Neuronal Excitation by Inhibitory GABA A Receptors. Science 1995, 269, 977–981.
- Viitanen, T.; Ruusuvuori, E.; Kaila, K.; Voipio, J. The K+-Cl−cotransporter KCC2 Promotes GABAergic Excitation in the Mature Rat Hippocampus. J. Physiol. 2010, 588, 1527–1540.
- Velazquez, J.L.P. Bicarbonate-Dependent Depolarizing Potentials in Pyramidal Cells and Interneurons during Epileptiform Activity. Eur. J. Neurosci. 2003, 18, 1337–1342.
- Rivera, C.; Voipio, J.; Kaila, K. Two Developmental Switches in GABAergic Signalling: The K+-Cl-cotransporter KCC2 and Carbonic Anhydrase CAVII. J. Physiol. 2005, 562, 27–36.
- Ruusuvuori, E.; Li, H.; Huttu, K.; Palva, J.M.; Smirnov, S.; Rivera, C.; Kaila, K.; Voipio, J. Carbonic Anhydrase Isoform VII Acts as a Molecular Switch in the Development of Synchronous Gamma-Frequency Firing of Hippocampal CA1 Pyramidal Cells. J. Neurosci. 2004, 24, 2699–2707.
- Lim, L.-L.; Foldvary, N.; Mascha, E.; Lee, J. Acetazolamide in Women with Catamenial Epilepsy. Epilepsia 2001, 42, 746–749.
- Reiss, W.G.; Oles, K.S. Acetazolamide in the Treatment of Seizures. Ann. Pharmacother. 1996, 30, 514–519.
- Hamidi, S.; Avoli, M. Carbonic Anhydrase Inhibition by Acetazolamide Reduces In Vitro Epileptiform Synchronization. Neuropharmacology 2015, 95, 377–387.
- Lillis, K.P.; Kramer, M.A.; Mertz, J.; Staley, K.J.; White, J.A. Pyramidal Cells Accumulate Chloride at Seizure Onset. Neurobiol. Dis. 2012, 47, 358–366.
- Woodbury, D.M.; Rollins, L.T.; Gardner, M.D.; Hirschi, W.L.; Hogan, J.R.; Rallison, M.L.; Tanner, G.S.; Brodie, D.A. Effects of Carbon Dioxide on Brain Excitability and Electrolytes. Am. J. Physiol. Content 1957, 192, 79–90.
- Maren, T.H. Carbonic Anhydrase: Chemistry, Physiology, and Inhibition. Physiol. Rev. 1967, 47, 595–781.
- Masuda, Y.; Utsui, Y.; Shiraishi, Y.; Karasawa, T.; Yoshida, K.; Shimizu, M. Relationships between Plasma Concentrations of Diphenylhydantoin, Phenobarbital, Carbamazepine, and 3-Sulfamoylmethyl-1,2-Benzisoxazole (AD-810), a New Anticonvulsant Agent, and Their Anticonvulsant or Neurotoxic Effects in Experimental Animals. Epilepsia 1979, 20, 623–633.
- Seizure and Epilepsy Medicines. Available online: https://www.epilepsy.com/learn/treating-seizures-and-epilepsy/seizure-and-epilepsy-medicines (accessed on 3 September 2021).
- Schauf, C. Zonisamide Enhances Slow Sodium Inactivation inMyxicola. Brain Res. 1987, 413, 185–188.
- Rock, D.M.; Macdonald, R.L.; Taylor, C.P. Blockade of Sustained Repetitive Action Potentials in Cultured Spinal Cord Neurons by Zonisamide (AD 810, CI 912), a Novel Anticonvulsant. Epilepsy Res. 1989, 3, 138–143.
- Suzuki, S.; Kawakami, K.; Nishimura, S.; Watanabe, Y.; Yagi, K.; Scino, M.; Miyamoto, K. Zonisamide blocks T-type Calcium Channel in Cultured Neurons of Rat Cerebral Cortex. Epilepsy Res. 1992, 12, 21–27.
- Ueda, Y.; Doi, T.; Tokumaru, J.; Willmore, L. Effect of zonisamide on molecular regulation of glutamate and GABA transporter proteins during epileptogenesis in rats with hippocampal seizures. Mol. Brain Res. 2003, 116, 1–6.
- Yamamura, S.; Saito, H.; Suzuki, N.; Kashimoto, S.; Hamaguchi, T.; Ohoyama, K.; Suzuki, D.; Kanehara, S.; Nakagawa, M.; Shiroyama, T.; et al. Effects of zonisamide on Neurotransmitter Release Associated with Inositol Triphosphate Receptors. Neurosci. Lett. 2009, 454, 91–96.
- Wilfong, A.A.; Willmore, L.J. Zonisamide? A Review of Experience and Use in Partial Seizures. Neuropsychiatr. Dis. Treat. 2006, 2, 269–280.
- Mori, A.; Noda, Y.; Packer, L. The Anticonvulsant Zonisamide Scavenges Free Radicals. Epilepsy Res. 1998, 30, 153–158.
- Shank, R.P.; Gardocki, J.F.; Vaught, J.L.; Davis, C.B.; Schupsky, J.J.; Raffa, R.B.; Dodgson, S.J.; Nortey, S.O.; Maryanoff, B.E. Topiramate: Preclinical Evaluation of a Structurally Novel Anticonvulsant. Epilepsia 1994, 35, 450–460.
- Wauquier, A.; Zhou, S. Topiramate: A Potent Anticonvulsant in the Amygdala-Kindled Rat. Epilepsy Res. 1996, 24, 73–77.
- Rigoulot, M.-A.; Boehrer, A.; Nehlig, A. Effects of Topiramate in Two Models of Genetically Determined Generalized Epilepsy, the GAERS and the Audiogenic Wistar AS. Epilepsia 2003, 44, 14–19.
- Chen, C.; Lang, S.; Xu, G.; Liu, X.; Zuo, P. Effects of Topiramate on Seizure Susceptibility in Kainate-Kindled Rats: Involvement of Peripheral-Type Benzodiazepine Receptors. Seizure 2008, 17, 358–363.
- Griffin, A.; Hamling, K.; Hong, S.; Anvar, M.; Lee, L.P.; Baraban, S.C. Preclinical Animal Models for Dravet Syndrome: Seizure Phenotypes, Comorbidities and Drug Screening. Front. Pharmacol. 2018, 9, 573.
- Chiron, C. Current Therapeutic Procedures in Dravet Syndrome. Dev. Med. Child Neurol. 2011, 53, 16–18.
- Lee, H.; Kim, D.W. Usefulness of Extended-Release Topiramate in Patients with Epilepsy: A Two-Year Retention Study. J. Clin. Pharm. Ther. 2021, 46, 1412–1417.
- Herrero, A.I.; Del Olmo, N.; González-Escalada, J.R.; Solís, J.M. Two New Actions of Topiramate: Inhibition of Depolarizing GABAA-Mediated Responses and Activation of a Potassium Conductance. Neuropharmacology 2002, 42, 210–220.
- Aribi, A.M.; Stringer, J.L. Effects of Antiepileptic Drugs on Extracellular pH Regulation in the Hippocampal CA1 Region In Vivo. Epilepsy Res. 2002, 49, 143–151.
- White, H.S.; Brown, S.D.; Woodhead, J.H.; Skeen, G.A.; Wolf, H.H. Topiramate Modulates GABA-evoked Currents in Murine Cortical Neurons By A Nonbenzodiazepine Mechanism. Epilepsia 2000, 41, 17–20.
- Petroff, O.A.C.; Hyder, F.; Rothman, D.L.; Mattson, R.H. Topiramate Rapidly Raises Brain GABA in Epilepsy Patients. Epilepsia 2001, 42, 543–548.
- Gryder, D.S.; Rogawski, M.A. Selective Antagonism of GluR5 Kainate-Receptor-Mediated Synaptic Currents by Topiramate in Rat Basolateral Amygdala Neurons. J. Neurosci. 2003, 23, 7069–7074.
- DeLorenzo, R.J.; Sombati, S.; Coulter, U.A. Effects of Topiramate on Sustained Repetitive Firing and Spontaneous Recurrent Seizure Discharges in Cultured Hippocampal Neurons. Epilepsia 2000, 41, 40–44.

