 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Elena Conte | + 3629 word(s) | 3629 | 2021-10-20 10:27:18 | | | |
| 2 | Jason Zhu | -28 word(s) | 3601 | 2021-11-01 03:14:57 | | | | |
| 3 | Jason Zhu | -2 word(s) | 3599 | 2021-11-03 02:26:43 | | |
Video Upload Options
Store-operated Ca 2+ entry (SOCE), also known as capacitive calcium entry (CCE), consists in a Ca2+influx downstream of ER/SR Ca2+stores and it is a pivotal mechanism in cellular calcium signaling and in maintaining cellular calcium homeostasis. The concept of SOCE was first postulated by Putney in 1986 who demonstrated that in salivary gland cells the depletion of internal Ca2+stores controlled the extent of Ca2+influx.
1. Introduction
In skeletal muscle fibers, intracellular Ca 2+ ions are essential signaling mediators that play a critical role in contraction and muscle plasticity mechanisms by regulating protein synthesis and degradation, fiber type shifting, calcium-regulated proteases and transcription factors and mitochondrial adaptations [1]. Ca 2+ homeostasis alteration has been observed in a growing number of muscle diseases, such as muscular hypotonia and myopathies [2][3][4], muscular dystrophies [5][6][7], cachexia [8] and age-related sarcopenia [9][10][11][12][13]. For this reason, the preservation of Ca 2+ homeostasis is an important and necessary requisite for maintaining skeletal muscle structure and function. Cellular Ca 2+ homeostasis is maintained through the precise and coordinated function of Ca 2+ transport molecules, Ca 2+ buffer/binding proteins such as calsequestrin or calreticulin, and several calcium channels. These include the plasma membrane calcium ATPases (PMCAs) that actively pump Ca 2+ out of the cell [14]; the Ca 2+ -release-activated-Ca 2+ (CRAC) channel located in the plasma membrane (PM) and activated by the endoplasmic/sarcoplasmic reticulum (ER/SR)-Ca 2+ release; and the sarco-/endoplasmic reticular calcium ATPase (SERCA) located in the ER/SR that transport Ca 2+ back into the ER/SR [15]. In skeletal muscle, calcium homeostasis is achieved when there is a balance between the calcium channels/buffers functions and three muscle principal systems: excitation–contraction (EC) coupling, excitation-coupled Ca 2+ entry (ECCE), and store-operated Ca 2+ entry (SOCE). EC coupling is the process mediated by mechanical coupling between the dihydropyridine receptor (DHPR) in the transverse tubule membrane, specialized invaginations of the sarcolemma, and the ryanodine receptor type 1 (RYR1) ion channel located in the ER/SR membrane. In this process, an action potential in the transverse tubule and the voltage-dependent conformational change of DHPR trigger the release of Ca 2 + from the sarcoplasmic reticulum to drive muscle contraction [16]. ECCE is a store-independent Ca 2+ entry pathway mediated by the DHPR, RYR1, and by a yet to be identified Ca 2+ entry channel with properties corresponding to those of store-operated Ca 2+ channels. It is triggered by sustained or repetitive depolarization and contributes to muscle contractility [17][18][19]. SOCE is a Ca 2+ -entry process activated by depletion of intracellular stores that contributes to the regulation of various functions in many cell types. It is mediated by the interaction between stromal-interacting molecule-1 (STIM1), the Ca 2+ sensor of ER/SR [20], and Orai1, the key CRAC channel located in the transverse tubules [21]. Aberrant SOCE can trigger a change of intracellular Ca 2+ signaling in skeletal muscle, thus causing or contributing to the pathogenesis of various skeletal muscle disorders. Therefore, therapies focused on restoring SOCE mechanism and targeting SOCE-associated proteins are promising for the treatment of SOCE-related skeletal muscle disorders.
2. Molecular Components of SOCE
STIM1 and STIM2 are characterized by a > 74% sequence similarity (66% sequence identity) between their key domains (EF/SAM domains, CC1, SOAR), but work differently as Ca 2+ sensors and activators of SOCE [22]. Although STIM2 is an analogue protein of STIM1, its functional role and contribution to the whole SOCE-mediated Ca 2+ signaling in skeletal muscle are not clear. An initial study on the role of STIM2 in SOCE demonstrated that STIM2 was a weaker Orai1 activator and a slow responder to ER luminal Ca 2+ changes compared to STIM1 [23]. Successively, Ong et al. reported that STIM2 is activated under a mild depletion of Ca 2+ stores and is able to form heterodimers with STIM1, thus increasing the recruitment of STIM1 to the ER/SR-PM junction and facilitating its activation [24]. A subsequent study showed that, in STIM2-knockdown mouse primary skeletal myotubes, STIM2 is able to interact with SERCA1a, causing a reduction of its activity during skeletal muscle contraction [25]. In addition, SOCE is significantly reduced after STIM2-knockdown, suggesting that STIM2 also contributes to SOCE in skeletal muscle [26]. Furthermore, STIM2 variants have different roles in the modulation of SOCE; STIM2.1 and STIM2.2 have been described to play as an inhibitor and an activator of SOCE, respectively, while the role of STIM2.3 still remains unclear [26].
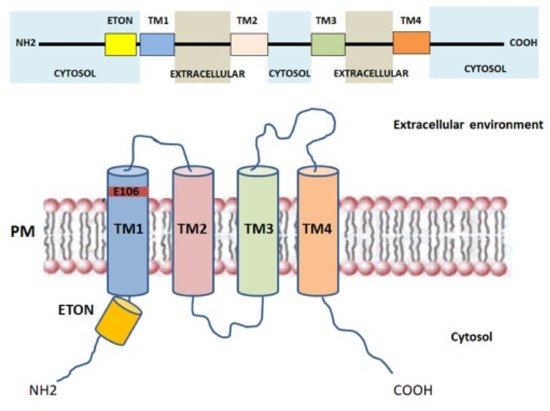
Orai proteins have been identified as key components of the Ca 2+ release-activated Ca 2+ channel (CRAC channel) [21][27] and are considered the major SOCE-mediating channels in skeletal muscle cells [28][29]. Particularly, ORAI (also called CRACM) proteins are located in the transverse tubules of PM and are responsible for the formation of the Ca 2+ -selective ion pores. Three Orai isoforms (Orai1-3, or CRACM1-3) encoded by homologous genes and two versions of Orai1, Orai1α and Orai1β, arising from alternative translation initiation [30], were identified in the human genome [31]. The presence of a point mutation (R91W) in Orai1, leading to loss of I CRAC current in human T cells, suggested the link between Orai1, in both Orai1α and β isoforms, and CRAC channel function [21][32][33][34]. Orai channels form hexameric complexes arranged around a central highly Ca 2+ -selective pore [35]. Each Orai subunit is composed of four transmembrane helices (TM1-TM4) connected by one intracellular (TM2-TM3) and two extracellular loops (TM1-TM2, TM3-TM4) with the N - and C -regions facing the cytoplasm that mediate the interaction with STIM1, STIM2, and other regulatory proteins [36] ( Figure 2 ). The Ca 2+ pore is formed by six TM1 domains surrounded by TM2-TM3, which provide stability to the structure [37], and by a cytosolic C -terminus. The glutamate at position 106, situated at the extracellular end of TM1, provides the binding site for Ca 2+ ions inside the channel and confers the high Ca 2+ selectivity to the CRAC channel [31][38]. Close to TM1 region, a conserved sequence called extended transmembrane Orai1 N -terminal (ETON) region is present. This region contributes to the interaction between the N -terminus of Orai1 and STIM1 [39]. Indeed, Orai1 mutants that lack the ETON region result in a reduced interaction with STIM1 [39]. The TM4 outer ring transduces the gating signal generated by the binding of STIM1 to the ORAI1 C -terminus [28][40]. Evidence demonstrates that the TM3-TM4 loop is pivotal for STIM1-mediated Orai1 gating [41] as it controls the interaction with Orai N -terminus and promotes a permissive conformation of Orai channel [42]. Orai1 gating depends on the folding state of STIM1 located in the ER/SR membrane. The interaction between Orai1 and STIM1 leads to conformational changes of the C -terminus in the Orai1 complex, that are subsequently transmitted to the pore region [43]. Furthermore, it has been shown that, in mouse skeletal myotubes, Orai1 was able to interact with Mitsugumin 53, a protein responsible for membrane repair of various cells, finally increasing Ca 2+ entry through Orai1 via a SOCE mechanism [44]. The physiological roles of Orai2 and Orai3 in skeletal muscle are still poorly defined. The biophysical properties of Orai2 and Orai3, including Ca 2+ permeation, are different from those of Orai1, suggesting that these proteins are little involved in SOCE [45]. Recently, Yoast et al. demonstrated that native Orai2-3 heteromerize with Orai1 to form a native CRAC channel regulating the activation of NFAT1 and NFAT4 isoforms. These results suggest that the levels of Orai2-3 relative to Orai1 can alter the magnitude of SOCE and downstream signaling [46].
In skeletal muscle, the non-selective transient receptor potential channels (TRPCs) have also been proposed as emerging SOCE channels [47]. Indeed, STIM1, ORAI1 and TRP channels act efficiently and are located near the triad junction [48]. TRPCs are activated downstream of the PLC/ IP3/DAG pathway; they contribute to the entry of extracellular Ca 2+ with moderate Ca 2+ selectivity [49]. TRP channels include seven isoforms (TRPC1-7), but only TRPC1 and TRPC3-6 are expressed in skeletal muscle [50][51]. TRPC consist of six transmembrane domains (TM1-TM6) with the TM5-TM6 loop forming the channel pore and with the N - and C -regions facing into the cytoplasm [52]. The cytosolic N -terminus contains an ankyrin repeat domain; while the cytosolic C -terminus contains a TRP-like domain for phosphatidylinositol biphosphate (PIP2) regulation necessary for channel activity and desensitization, a calmodulin/inositol 1,4,5-trisphosphate receptor-binding (CIRB) site and a Ca 2+ binding EF-hand [53] ( Figure 3 A). Among all skeletal muscle isoforms, the best characterized is TRPC1, which has a role in Ca 2+ homeostasis during sustained contractile muscle activity and is considered the best candidate to conduct Ca 2+ influx during SOCE. TRPC1, which acts as a SOCE channel, participates in myoblasts migration and fusion via calpain activation during the terminal differentiation process [53]. Indeed, Antigny and colleagues demonstrated that TRPC1 and TRPC4-mediated Ca 2+ entry was important to produce normal-sized myotubes. Conversely, overexpression of STIM1 with TRPC4 or TRPC1 increased SOCE, accelerated myoblast fusion, and produced hypertrophic myotubes [54]. Additionally, the Ca 2+ entry through TRPC1 has been shown to play a role in myoblast differentiation and in muscle regeneration by modulating the PI3K/Akt/mTOR/p70S6K pathway. In fact, in myoblasts derived from Trpc1 −/− mice, the phosphorylation of both Akt and p70S6K proteins, as well as the activation of PI3K, were inhibited. This evidence suggests that Ca 2+ entry through TRPC1 plays a role in the activation of this pathway [55]. TRPC1 or TRPC4 has been shown to complex with STIM1 and STIM1L, thus contributing to the activation of skeletal muscle SOCE associated with sustained Ca 2+ transients during repetitive membrane depolarizations. This process, in turn, promotes myogenesis and maintains a fast repetitive Ca 2+ release in human skeletal myotubes [56]. TRPC1 seems to be activated by both STIM1 isoforms, with STIM1L more efficient in activating TRPC1than STIM1 [56][22]. Furthermore, recent studies reported that the TRPC1 mediated currents did not reproduce the biophysical properties of I CRAC [57], defining TRPC1 current as I soc . However, this observation does not rule out the TRPC channels’ involvement in SOCE. Experimental studies have shown that STIM1 interacts with and directly activates TRPC1 channels not only by binding through its SOAR region with TRPC1, but also through electrostatic interactions between highly positively charged lysines located in the STIM1 lysine-rich domain and the TRPC1 aspartate residues [47][58]. STIM1 residues involved in TRPC1 gating are distinct from those involved in Orai1 activation. In addition, a different mechanism of TRPCs versus Orai channels by STIM1 is suggested [58]. However, activation of TRPC1 requires an additional crucial functional interaction with Orai1. This observation was confirmed by data showing that Orai1-mediated Ca 2+ entry triggers the recruitment of TRPC1 into the PM where it is then activated by STIM1 [59][60]. In human skeletal muscle the expression of TRPC3 and TRPC6 isoforms is firmly established, and it has been found that Orai1 is able to bind to the N - and C - terminals of TRPC3 or TRPC6. Actually, it is not clear if they are able to act as SOCE channels without Orai1 interaction [61]. Furthermore, in skeletal muscle, STIM1L, like Orai1, is known to interact with TRPC3 and TRPC6 [56][62], but it is not clear whether this interaction is fundamental for SOCE mechanism.
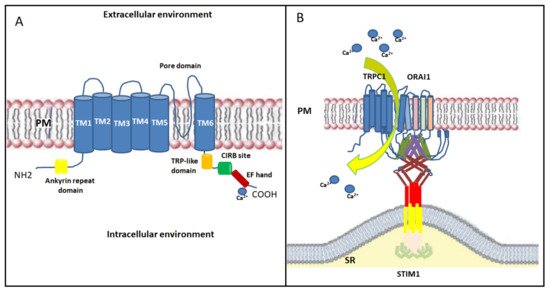
Finally, the overall findings on STIM1, Orai1 and TRPC suggest the formation of a dynamic STIM1-Orai1-TRPC1 ternary complex as the SOC channels; after store depletion, STIM1 oligomers transfer to the ER/SR-PM junctions where they bind Orai1 and TRPC1 in lipid raft domains, activating both Ca 2+ channels [57] ( Figure 3 B). Further studies on the relationships between TRPCs, Orais and STIM1 are needed to examine this and other scenarios.
3. SOCE Mechanism in Skeletal Muscle
SOCE, also known as capacitive calcium entry (CCE), consists in a Ca 2+ influx downstream of ER/ SR Ca 2+ stores and it is a pivotal mechanism in cellular calcium signaling and in maintaining cellular calcium homeostasis [63]. The concept of SOCE was first postulated by Putney in 1986 who demonstrated that in salivary gland cells the depletion of internal Ca 2+ stores controlled the extent of Ca 2+ influx [64]. Successively, in 2001, SOCE was identified in skeletal muscle fibers of adult mice for the first time [65]. Generally, SOCE is activated in response to the emptying of the SR/ER-Ca 2+ stores and is followed by a limited increase in [Ca 2+ ] i due to the smaller SR/ER volume. Two major families of intracellular Ca 2+ channels are responsible for linking extracellular stimuli to the release of Ca 2+ from the ER/SR: RyRs, which are opened by their direct interaction with the voltage-sensing dihydropyridine receptor in the PM, and the inositol 1,4,5-trisphosphate receptors (IP3R). In the second case, depletion of the ER/SR store essentially occurs following the stimulation of plasma membrane G protein-coupled receptors (GPCRs) through the activation of phospholipase C (PLC) subtypes and the related release of inositol 1,4,5-triphosphate (IP3); IP3 binds to the IP3R on the ER/SR membrane, triggering Ca 2+ release from the ER/SR lumen into the cytosol [66]. Experimentally, thapsigargin, cyclopiazonic acid and 2,5-di-(tert-butyl)-1,4-benzohydroquinone associated with a RYR1-activator (such as caffeine or KCl) are the most commonly used pharmacological tools for inhibiting the ER/SR SERCA pump and depleting ER/SR Ca 2+ independently of receptors and IP3 [67][68][69]. However, following ER/ SR Ca 2+ store depletion, a secondary influx of extracellular Ca 2+ through CRAC channels in the plasma membrane results in a more sustained increase in cytosolic Ca 2+ levels. Different SOCE models in skeletal muscle have been proposed, including the interaction between STIM1 and Orai1 [47], STIM1 and transient receptor potential canonical channels (TRPCs) [58], and STIM1-Orai1-TRPCs [57]. Among these, the direct coupling of ER-resident STIM1 to plasma membrane-resident Orai1 is currently considered as the most straightforward SOCE mechanism, and it has been studied in depth.
In the resting state, STIM1 is a dimer associated with its regulator SOCE-associated regulatory factor (SARAF), a single-pass transmembrane protein [70]. The process begins when depletion of Ca 2+ from the ER/SR leads to Ca 2+ dissociation from the luminal STIM1 N -terminal EF-hand domains. This determines the beginning of dramatic conformational changes in STIM1 that lead to the formation of an unfolded STIM1 protein and more closely associated luminal EF-hand domains. Unfolding and activation of STIM1 upon Ca 2+ dissociation causes SARAF dissociation [70] and STIM1 oligomerization [71] by associating the paired EF-SAM domains [72] and by the exposition of the STIM1-Orai1 activating region domain (SOAR domain) [73]. Successively, STIM1 oligomers migrate towards ER/SR regions juxtaposed to the PM and bind to it through the interaction between the poly-lys rich domain near the STIM1 C -terminus and the PM phospholipids [71], resulting in the “puncta” formation [74]. At the same time, rearrangements of some portions of the ER/SR towards the PM favor STIM1 interaction and puncta formation [75]. After puncta formation, STIM1 oligomers are able to bind with the N - and C - termini of Orai1 through SOAR region and activate Orai1 channels for Ca 2+ entry from the extracellular environment [63][74][76][77] ( Figure 4 ). It is well recognized that STIM1 is a phosphoprotein [78] and biochemical studies strongly contributed to understand the signaling responsible for the correct interaction between STIM1 and Orai1, and consequently for the correct functioning of SOCE. Particularly, Yazbeck et al. showed that STIM1 could be modulated by a Pyk2-dependent tyrosine phosphorylation at Y361 within the SOAR domain. This seems to be a critical step in activating Ca 2+ entry through Orai1 channels since it is required for Orai1 recruitment into STIM1 puncta and for STIM1-Orai1 interaction [79]. Furthermore, Lopez et al. showed that STIM1 phosphorylation at Y316 could enhance the formation of the CRAC signaling complex, which contribute to SARAF dissociation from STIM1 and regulation of slow Ca 2+ -dependent inactivation [70].
Another hypothesis on the SOCE mechanism postulates that, in skeletal muscle, STIM1 and Orai1 pre-localize under resting conditions within the triad junction, a specialized macrostructure composed of a parallel transverse tubule and two opposing ER/SR membranes. They remain inactive until ER/SR depletion triggers conformational changes in STIM1 and direct activation of Orai1-mediated Ca 2+ influx [63]; this allows an extremely fast and efficient trans-sarcolemmal Ca 2+ influx during store depletion. Accordingly, in skeletal muscle, SOCE occurs in less than a second, i.e., significantly faster than in other types of cells where it can require up to several seconds [80].
The precise stoichiometry of the STIM1-Orai1 complex that forms the functional core of the CRAC channel still needs clarification and it has long been a subject of debate [81]. Several studies hypothesized that a dimer of STIM1s binds to a pair of Orai1 C -terminal fragments (in a 1:1 STIM1: Orai1 stoichiometry) [82][83][84]. Alternatively, each dimer interacts with only a single C -terminal tail, leaving the remaining STIM1 subunit free to cross-link with a different Orai1 channel (two STIM1 molecules around a single Orai1 channel, in a 2:1 STIM1: Orai1 stoichiometry) [85]. More recently, it has been reported that the native SOCE complex includes only a few STIM1 dimers associated with a single Orai1 channel [86].
4. Therapeutic Perspectives for Counteracting SOCE-Related Skeletal Muscle Diseases
As knowledge about the role of SOCE in skeletal muscle diseases accumulates, there has been a growing interest in developing molecules targeting SOCE and identifying therapies that can be used for specific treatments. Indeed, several studies recently aimed to develop SOCE modulators to reduce SOCE activation following the pathological skeletal muscle GoF mutations mentioned above. For example, Rahaman and colleagues used in silico screening to identify FDA-approved drugs able to suppress the SOCE mechanism. Particularly, leflunomide and teriflunomide, FDA-approved drugs for the treatment of rheumatoid/psoriatic arthritis and multiple sclerosis, respectively, were able to inhibit SOCE at therapeutically-achievable concentrations; furthermore, lansoprazole, tolvaptan and roflumilast resulted in even more selective molecules to suppress the SOCE mechanism [87]. Recently, a variety of new small molecules blocking CRAC channels have been identified and developed, such as pyrtriazoles or pyrazole SKF-96365 analogues [88][89]. However, all currently available SOCE inhibitors show no specific effects [90][91].
Regarding dystrophies, and DMD in particular, at present there are no effective treatments and the glucocorticoids which act as anti-inflammatory agents are often used to stop progressive muscle damage [92][93]. Prednisone, prednisolone, and deflazacort, mostly through inhibition of NF-κB signaling, represent a gold standard for the treatment of DMD for their ability to exert long-term protective effects [94]. Importantly, to date, an increasing variety of therapeutic strategies aimed at restoring dystrophin production and to preserve muscle mass has been proposed, ranging from gene therapy to antisense oligonucleotide therapies [95][96]. Several studies propose therapeutic approaches for DMD aimed not only at restoring dystrophin function but also to mitigate secondary and downstream pathological mechanisms that contribute to the disease’s progression, such as calcium dysregulation, oxidative stress, mitochondria dysfunction, fibrosis and muscle wasting. Among all, since increased calcium concentration plays a significant role in the pathogenesis of DMD, therapeutic strategies aimed at controlling Ca 2+ are in progress. The spider venom toxin AT-300/GsMTx4, a peptide that blocks the mechanosensitive Ca 2+ channels, for example, prevented the rise of intracellular resting Ca 2+ with modest benefits in mdx mice [97]. Another therapeutic option is treatment with the small drug ARM210/S48168, a ryanodine channel complex stabilizer, which improves muscle functionality in mdx mice, notably in the diaphragm [7]. Although SOCE increase in DMD is known, little evidence demonstrates that this alteration is linked to an increase in the STIM1/Orai1/TRPC expression. In this context, STIM1/Orai1/TRPC proteins may represent valuable therapeutic targets for testing compounds/drugs that regulate Ca 2+ signal alteration in DMD, and focused studies in this field are highly desirable.
Finally, regarding skeletal muscle wasting disorders, knowledge of the real role of the SOCE mechanism, in particular during cachexia and aged-sarcopenia, is a fundamental requirement for finding a potential therapy. Nutrition is a key factor for the therapy of these conditions because both the quality and quantity of nutrients are pivotal for improving muscle anabolism, reducing catabolism, and lightening the prognosis [98]. However, although nutrition alone can prevent or minimize further skeletal muscle loss, it cannot completely reverse these conditions. For this reason, for example for cachexia, a multifactorial approach is currently proposed [99]. In this respect, a potential therapeutic option for cancer cachexia syndrome is represented by growth hormone secretagogues (GHS) [100][101], ghrelin mimetics known to increase appetite, lean and fat mass [102]. Recently, it was shown that GHS administration, in particular the well-known peptidyl GHS hexarelin and a novel peptidomimetic GHS JMV 2894, efficaciously prevented Ca 2+ homeostasis alteration and SOCE decrease in skeletal muscle of cachectic rats [8]. Interestingly, JMV2894 was able to restore STIM1 and ORAI1 gene expression [8]. A direct interference of JMV2894 with SOCE mechanism is not excluded. Indeed, given the small molecular size of JMV2894, an interaction with the RyR protein and a consequent stabilizer activity could be postulated. This is also supported by the positive effects observed regarding SR responsiveness to caffeine, demonstrated in JMV2894 treated rats [8]. All these findings demonstrate that SOCE activity strongly contributes to the dysregulation of Ca 2+ homeostasis observed in the cachectic muscles suggesting that SOCE could be considered a potential target for cachexia therapy. Likewise, sarcopenia cannot be completely reversed by conventional nutritional support and/or increased physical activity, and SOCE could be considered a potential biomarker and target for therapeutical interventions for prevention or for counteracting sarcopenia. To achieve this goal, additional focused studies are still needed. In this context, the evaluation of senolytics and senostatics drugs, molecules considered to be revolutionizing in the field of aging research [103], on the SOCE mechanism could be very appealing.
References
- Gehlert, S.; Bloch, W.; Suhr, F. Ca2+ dependent regulations and signaling in skeletal muscle: From electro-mechanical coupling. Int. J. Mol. Sci. 2015, 16, 1066–1095.
- Böhm, J.; Bulla, M.; Urquhart, J.E.; Malfatti, E.; Williams, S.G.; O’Sullivan, J.; Szlauer, A.; Koch, C.; Baranello, G.; Mora, M.; et al. ORAI1 Mutations with Distinct Channel Gating Defects in Tubular Aggregate Myopathy. Hum. Mutat. 2017, 38, 426–438.
- Silva-Rojas, R.; Laporte, J.; Böhm, J. STIM1/ ORAI1 Loss-of-Function and Gain-of-Function Mutations Inversely Impact on SOCE and Calcium Homeostasis and Cause Multi-Systemic Mirror Diseases. Front. Physiol. 2020, 11, 604941.
- Conte, E.; Pannunzio, A.; Imbrici, P.; Camerino, G.M.; Maggi, L.; Mora, M.; Gibertini, S.; Cappellari, O.; De Luca, A.; Coluccia, M.; et al. Gain-of-Function STIM1 L96V Mutation Causes Myogenesis Alteration in Muscle Cells From a Patient Affected by Tubular Aggregate Myopathy. Front. Cell Dev. Biol. 2021, 9, 635063.
- Zhao, X.; Moloughney, J.G.; Zhang, S.; Komazaki, S.; Weisleder, N. Orai1 mediates exacerbated Ca(2+) entry in dystrophic skeletal muscle. PLoS ONE 2012, 7, e49862.
- Fraysse, B.; Liantonio, A.; Cetrone, M.; Burdi, R.; Pierno, S.; Frigeri, A.; Pisoni, M.; Camerino, C.; De Luca, A. The alteration of calcium homeostasis in adult dystrophic mdx muscle fibers is worsened by a chronic exercise in vivo. Neurobiol. Dis. 2004, 17, 144–154.
- Capogrosso, R.F.; Mantuano, P.; Uaesoontrachoon, K.; Cozzoli, A.; Giustino, A.; Dow, T.; Srinivassane, S.; Filipovic, M.; Bell, C.; Vandermeulen, J.; et al. Ryanodine channel complex stabilizer compound S48168/ARM210 as a disease modifier in dystrophin-deficient mdx mice: Proof-of-concept study and independent validation of efficacy. FASEB J. 2018, 32, 1025–1043.
- Conte, E.; Camerino, G.M.; Mele, A.; De Bellis, M.; Pierno, S.; Rana, F.; Fonzino, A.; Caloiero, R.; Rizzi, L.; Bresciani, E.; et al. Growth hormone secretagogues prevent dysregulation of skeletal muscle calcium homeostasis in a rat model of cisplatin-induced cachexia. J. Cachexia Sarcopenia Muscle 2017, 8, 386–404.
- Weisleder, N.; Brotto, M.; Komazaki, S.; Pan, Z.; Zhao, X.; Nosek, T.; Parness, J.; Takeshima, H.; Ma, J. Muscle aging is associated with compromised Ca2+ spark signaling and segregated intracellular Ca2+ release. J. Cell Biol. 2006, 174, 639–645.
- Fraysse, B.; Desaphy, J.F.; Rolland, J.F.; Pierno, S.; Liantonio, A.; Giannuzzi, V.; Camerino, C.; Didonna, M.P.; Cocchi, D.; De Luca, A.; et al. Fiber type-related changes in rat skeletal muscle calcium homeostasis during aging and restoration by growth hormone. Neurobiol. Dis. 2006, 21, 372–380.
- Romero-Suarez, S.; Shen, J.; Brotto, L.; Hall, T.; Mo, C.; Valdivia, H.H.; Andresen, J.; Wacker, M.; Nosek, T.M.; Qu, C.K.; et al. Muscle-specific inositide phosphatase (MIP/MTMR14) is reduced with age and its loss accelerates skeletal muscle aging process by altering calcium homeostasis. Aging 2010, 2, 504–513.
- Andersson, D.C.; Betzenhauser, M.J.; Reiken, S.; Meli, A.C.; Umanskaya, A.; Xie, W.; Shiomi, T.; Zalk, R.; Lacampagne, A.; Marks, A.R. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab. 2011, 14, 196–207.
- Thornton, A.M.; Zhao, X.; Weisleder, N.; Brotto, L.S.; Bougoin, S.; Nosek, T.M.; Reid, M.; Hardin, B.; Pan, Z.; Ma, J.; et al. Store-operated Ca(2+) entry (SOCE) contributes to normal skeletal muscle contractility in young but not in aged skeletal muscle. Aging 2011, 3, 621–634.
- Brini, M.; Cali, T.; Ottolini, D.; Carafoli, E. The plasma membrane calcium pump in health and disease. FEBS J. 2013, 280, 5385–5397.
- Toyoshima, C. How Ca2+-ATPase pumps ions across the sarcoplasmic reticulum membrane. Biochim. Biophys. Acta 2009, 1793, 943–946.
- Kuo, I.Y.; Ehrlich, B.E. Signaling in Muscle Contraction. Cold Spring Harb. Perspect. Biol. 2015, 7, a006023.
- Cherednichenko, G.; Hurne, A.M.; Fessenden, J.D.; Lee, E.H.; Allen, P.D.; Beam, K.G.; Pessah, I.N. Conformational activation of Ca2+ entry by depolarization of skeletal myotubes. Proc. Natl. Acad. Sci. USA 2004, 101, 15793–15798.
- Dirksen, R.T. Checking your SOCCs and feet: The molecular mechanisms of Ca2+ entry in skeletal muscle. J. Physiol. 2009, 587 Pt 13, 3139–3147.
- Bannister, R.A.; Pessah, I.N.; Beam, K.G. The skeletal L-type Ca(2+) current is a major contributor to excitation-coupled Ca(2+) entry. J. Gen. Physiol. 2009, 133, 79–91.
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445.
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185.
- Stathopulos, P.B.; Ikura, M. Structure and function of endoplasmic reticulum STIM calcium sensors. Curr. Top. Membr. 2013, 71, 59–93.
- Bird, G.S.; Hwang, S.Y.; Smyth, J.T.; Fukushima, M.; Boyles, R.R.; Putney, J.W., Jr. STIM1 is a calcium sensor specialized for digital signaling. Curr. Biol. 2009, 19, 1724–1729.
- Ong, H.L.; de Souza, L.B.; Zheng, C.; Cheng, K.T.; Liu, X.; Goldsmith, C.M.; Feske, S.; Ambudkar, I.S. STIM2 enhances receptor-stimulated Ca2+ signaling by promoting recruitment of STIM1 to the endoplasmic reticulum-plasma membrane junctions. Sci. Signal. 2015, 8, ra3.
- Oh, M.R.; Lee, K.J.; Huang, M.; Kim, J.O.; Kim, D.H.; Cho, C.H.; Lee, E.H. STIM2 regulates both intracellular Ca(2+) distribution and Ca(2+) movement in skeletal myotubes. Sci. Rep. 2017, 7, 17936.
- Rana, A.; Yen, M.; Sadaghiani, A.M.; Malmersjo, S.; Park, C.Y.; Dolmetsch, R.E.; Lewis, R.S. Alternative splicing converts STIM2 from an activator to an inhibitor of store-operated calcium channels. J. Cell Biol. 2015, 209, 653–669.
- Vig, M.; Peinelt, C.; Beck, A.; Koomoa, D.L.; Rabah, D.; Koblan-Huberson, M.; Kraft, S.; Turner, H.; Fleig, A.; Penner, R.; et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 2006, 312, 1220–1223.
- Wei-Lapierre, L.; Carrell, E.M.; Boncompagni, S.; Protasi, F.; Dirksen, R.T. Orai1-dependent calcium entry promotes skeletal muscle growth and limits fatigue. Nat. Commun. 2013, 4, 2805.
- Sztretye, M.; Geyer, N.; Vincze, J.; Al-Gaadi, D.; Olah, T.; Szentesi, P.; Kis, G.; Antal, M.; Balatoni, I.; Csernoch, L.; et al. SOCE Is Important for Maintaining Sarcoplasmic Calcium Content and Release in Skeletal Muscle Fibers. Biophys. J. 2017, 113, 2250–2496.
- Fukushima, M.; Tomita, T.; Janoshazi, A.; Putney, J.W. Alternative translation initiation gives rise to two isoforms of Orai1 with distinct plasma membrane mobilities. J. Cell Sci. 2012, 125 Pt 18, 4354–4361.
- Hou, X.; Pedi, L.; Diver, M.M.; Long, S.B. Crystal structure of the calcium release-activated calcium channel Orai. Science 2012, 338, 1308–1313.
- Feske, S.; Prakriya, M.; Rao, A.; Lewis, R. A severe defect in CRAC Ca2+ channel activation and altered K+ channel gating in T cells from immunodeficient patients. J. Exp. Med. 2005, 202, 651–662.
- Soboloff, J.; Spassova, M.; Tang, X.; Hewavitharana, T.; Xu, W.; Gill, D. Orai1 and STIM reconstitute store-operated calcium channel function. J. Biol. Chem. 2006, 281, 20661–20665.
- Yamashita, M.; Navarro-Borelly, L.; McNally, B.; Prakriya, M. Orai1 mutations alter ion permeation and Ca2+-dependent fast inactivation of CRAC channels: Evidence for coupling of permeation and gating. J. Gen. Physiol. 2007, 130, 525–540.
- Rothberg, B.S.; Wang, Y.; Gill, D.L. Orai channel pore properties and gating by STIM: Implications from the Orai crystal structure. Sci. Signal. 2013, 6, pe9.
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006, 443, 230–233.
- Amcheslavsky, A.; Wood, M.L.; Yeromin, A.V.; Parker, I.; Freites, J.A.; Tobias, D.J.; Cahalan, M.D. Molecular biophysics of Orai store-operated Ca2+ channels. Biophys. J. 2015, 108, 237–246.
- Zhou, Y.; Ramachandran, S.; Oh-Hora, M.; Rao, A.; Hogan, P.G. Pore architecture of the ORAI1 store-operated calcium channel. Proc. Natl. Acad. Sci. USA 2010, 107, 4896–4901.
- Derler, I.; Plenk, P.; Fahrner, M.; Muik, M.; Jardin, I.; Schindl, R.; Gruber, H.J.; Groschner, K.; Romanin, C. The extended transmembrane Orai1 N-terminal (ETON) region combines binding interface and gate for Orai1 activation by STIM1. J. Biol. Chem. 2013, 288, 29025–29034.
- Calloway, N.; Holowka, D.; Baird, B. A basic sequence in STIM1 promotes Ca2+ influx by interacting with the C-terminal acidic coiled coil of Orai1. Biochemistry 2010, 49, 1067–1071.
- Butorac, C.; Muik, M.; Derler, I.; Stadlbauer, M.; Lunz, V.; Krizova, A.; Lindinger, S.; Schober, R.; Frischauf, I.; Bhardwaj, R.; et al. A novel STIM1-Orai1 gating interface essential for CRAC channel activation. Cell Calcium 2019, 79, 57–67.
- Fahrner, M.; Pandey, S.K.; Muik, M.; Traxler, L.; Butorac, C.; Stadlbauer, M.; Zayats, V.; Krizova, A.; Plenk, P.; Frischauf, I.; et al. Communication between N terminus and loop2 tunes Orai activation. J. Biol. Chem. 2018, 293, 1271–1285.
- Derler, I.; Butorac, C.; Krizova, A.; Stadlbauer, M.; Muik, M.; Fahrner, M.; Frischauf, I.; Romanin, C. Authentic CRAC channel activityrequires STIM1 and the conserved portion of the Orai N terminus. J. Biol. Chem. 2018, 293, 1259–1270.
- Ahn, M.K.; Lee, K.J.; Cai, C.; Huang, M.; Cho, C.H.; Ma, J.; Lee, E.H. Mitsugumin 53 regulates extracellular Ca(2+) entry and intracellular Ca(2+) release via Orai1 and RyR1 in skeletal muscle. Sci. Rep. 2016, 6, 36909.
- Hoth, M.; Niemeyer, B.A. The neglected CRAC proteins: Orai2, Orai3, and STIM2. Curr. Top. Membr. 2013, 71, 237–271.
- Yoast, R.E.; Emrich, S.M.; Zhang, X.; Xin, P.; Johnson, M.T.; Fike, A.J.; Walter, V.; Hempel, N.; Yule, D.I.; Sneyd, J.; et al. The native ORAI channel trio underlies the diversity of Ca 2+ signaling events. Nat. Commun. 2020, 11, 2444.
- Yuan, J.P.; Zeng, W.; Huang, G.N.; Worley, P.F.; Muallem, S. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat. Cell Biol. 2007, 9, 636–645.
- Kiviluoto, S.; Decuypere, J.P.; De Smedt, H.; Missiaen, L.; Parys, J.B.; Bultynck, G. STIM1 as a key regulator for Ca2+ homeostasis in skeletal-muscle development and function. Skelet Muscle 2011, 1, 16.
- Gees, M.; Colsoul, B.; Nilius, B. The Role of Transient Receptor Potential Cation Channels in Ca2+ Signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a003962.
- Cheung, K.K.; Yeung, S.S.; Au, S.W.; Lam, L.S.; Dai, Z.Q.; Li, Y.H.; Yeung, E.W. Expression and association of TRPC1 with TRPC3 during skeletal myogenesis. Muscle Nerve 2011, 44, 358–365.
- Kunert-Keil, C.; Bisping, F.; Krüger, J.; Brinkmeier, H. Tissue-specific expression of TRP channel genes in the mouse and its variation in three different mouse strains. BMC Genom. 2006, 7, 159.
- Hellmich, U.A.; Gaudet, R. Structural biology of TRP channels. Handb. Exp. Pharmacol. 2014, 223, 963–990.
- Louis, M.; Zanou, N.; Van Schoor, M.; Gailly, P. TRPC1 regulates skeletal myoblast migration and differentiation. J. Cell Sci. 2008, 121, 3951–3959.
- Antigny, F.; Koenig, S.; Bernheim, L.; Frieden, M. During post-natal human myogenesis, normal myotube size requires TRPC1- and TRPC4-mediated Ca2+ entry. J. Cell Sci. 2013, 126 Pt 11, 2525–2533.
- Zanou, N.; Schakman, O.; Louis, P.; Ruegg, U.T.; Dietrich, A.; Birnbaumer, L.; Gailly, P. Trpc1 ion channel modulates phosphatidylinositol 3-kinase/Akt pathway during myoblast differentiation and muscle regeneration. J. Biol. Chem. 2012, 287, 14524–14534.
- Antigny, F.; Sabourin, J.; Sauc, S.; Bernheim, L.; Koenig, S.; Frieden, M. TRPC1 and TRPC4 channels functionally interact with STIM1L to promote myogenesis and maintain fast repetitive Ca(2+) release in human myotubes. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 806–813.
- Ong, H.L.; Cheng, K.T.; Liu, X.; Bandyopadhyay, B.C.; Paria, B.C.; Soboloff, J.; Pani, B.; Gwack, Y.; Srikanth, S.; Singh, B.B.; et al. Dynamic assembly of TRPC1-STIM1-Orai1 ternary complex is involved in store-operated calcium influx. Evidence for similarities in store-operated and calcium release-activated calcium channel components. J. Biol. Chem. 2007, 282, 9105–9116.
- Zeng, W.; Yuan, J.P.; Kim, M.S.; Choi, Y.J.; Huang, G.N.; Worley, P.F.; Muallem, S. STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol. Cell. 2008, 32, 439–448.
- Kim, M.S.; Zeng, W.; Yuan, J.P.; Shin, D.M.; Worley, P.F.; Muallem, S. Native store-operated Ca2+ influx requires the channel function of orai1 and TRPC1. J. Biol. Chem. 2009, 284, 9733–9741.
- Cheng, K.T.; Liu, X.; Ong, H.L.; Ambudkar, I.S. Functional requirement for Orai1 in store-operated TRPC1-STIM1 channels. J. Biol. Chem. 2008, 283, 12935–12940.
- Liao, Y.; Erxleben, C.; Yildirim, E.; Abramowitz, J.; Armstrong, D.L.; Birnbaumer, L. Orai proteins interact with TRPC channels and confer responsiveness to store depletion. Proc. Natl. Acad. Sci. USA 2007, 104, 4682–4687.
- Horinouchi, T.; Higashi, T.; Higa, T.; Terada, K.; Mai, Y.; Aoyagi, H.; Hatate, C.; Nepal, P.; Horiguchi, M.; Harada, T.; et al. Different binding property of STIM1 and its novel splice variant STIM1L to Orai1, TRPC3, and TRPC6 channels. Biochem. Biophys. Res. Commun. 2012, 428, 252–258.
- Stiber, J.; Hawkins, A.; Zhang, Z.S.; Wang, S.; Burch, J.; Graham, V.; Rosenberg, P. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat. Cell Biol. 2008, 10, 688–697.
- Putney, J.W., Jr. A model for receptor-regulated calcium entry. Cell Calcium 1986, 7, 1–12.
- Kurebayashi, N.; Ogawa, Y. Depletion of Ca2+ in the sarcoplasmic reticulum stimulates Ca2+ entry into mouse skeletal muscle fibres. J. Physiol. 2001, 533, 185–199.
- Venkatachalam, K.; van Rossum, D.B.; Patterson, R.L.; Ma, H.T.; Gill, D.L. The cellular and molecular basis of store-operated calcium entry. Nat. Cell Biol. 2002, 4, E263–E272.
- Des Georges, A.; Clarke, O.B.; Zalk, R.; Yuan, Q.; Condon, K.J.; Grassucci, R.A.; Hendrickson, W.A.; Marks, A.R.; Frank, J. Structural Basis for Gating and Activation of RyR1. Cell 2016, 167, 145–157.e17.
- Thastrup, O.; Cullen, P.J.; Drøbak, B.K.; Hanley, M.R.; Dawson, A.P. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc. Natl. Acad. Sci. USA 1990, 87, 2466–2470.
- Putney, J.W., Jr. Pharmacology of capacitative calcium entry. Mol. Interv. 2001, 1, 84–94.
- Lopez, E.; Frischauf, I.; Jardin, I.; Derler, I.; Muik, M.; Cantonero, C.; Salido, G.M.; Smani, T.; Rosado, J.A.; Redondo, P.C. STIM1 phosphorylation at Y 316 modulates its interaction with SARAF and the activation of SOCE and I CRAC. J. Cell Sci. 2019, 132, jcs226019.
- Luik, R.M.; Wang, B.; Prakriya, M.; Wu, M.M.; Lewis, R.S. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature 2008, 454, 538–542.
- Stathopulos, P.; Li, G.; Plevin, M.; Ames, J.; Ikura, M. Stored Ca2+ depletion-induced oligomerization of STIM1 via the EF-SAM region: An initiation mechanism for capacitive Ca2+ entry. J. Biol. Chem. 2006, 281, 35855–35862.
- Yuan, J.P.; Zeng, W.; Dorwart, M.R.; Choi, Y.J.; Worley, P.F.; Muallem, S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 2009, 11, 337–343.
- Shim, A.H.; Tirado-Lee, L.; Prakriya, M. Structural and functional mechanisms of CRAC channel regulation. J. Mol. Biol. 2015, 427, 77–93.
- Luik, R.M.; Wu, M.M.; Buchanan, J.; Lewis, R.S. The elementary unit of store-operated Ca2+ entry: Local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J. Cell Biol. 2006, 174, 815–825.
- Park, C.Y.; Hoover, P.J.; Mullins, F.M.; Bachhawat, P.; Covington, E.D.; Raunser, S.; Walz, T.; Garcia, K.C.; Dolmetsch, R.E.; Lewis, R.S. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 2009, 136, 876–890.
- Zhou, Y.; Cai, X.; Nwokonko, R.M.; Loktionova, N.A.; Wang, Y.; Gill, D.L. The STIM-Orai coupling interface and gating of the Orai1 channel. Cell Calcium 2017, 63, 8–13.
- Williams, R.T.; Manji, S.S.; Parker, N.J.; Hancock, M.S.; Van Stekelenburg, L.; Eid, J.P.; Senior, P.V.; Kazenwadel, J.S.; Shandala, T.; Saint, R.; et al. Identification and characterization of the STIM (stromal interaction molecule) gene family: Coding for a novel class of transmembrane proteins. Biochem. J. 2001, 357 Pt 3, 673–685.
- Yazbeck, P.; Tauseef, M.; Kruse, K.; Amin, M.R.; Sheikh, R.; Feske, S.; Komarova, Y.; Mehta, D. STIM1 Phosphorylation at Y361 Recruits Orai1 to STIM1 Puncta and Induces Ca 2+ Entry. Sci. Rep. 2017, 7, 42758.
- Edwards, J.N.; Friedrich, O.; Cully, T.R.; von Wegner, F.; Murphy, R.M.; Launikonis, B.S. Upregulation of store-operated Ca2+ entry in dystrophic mdx mouse muscle. Am. J. Physiol. Cell Physiol. 2010, 299, C42–C50.
- Yen, M.; Lewis, R.S. Numbers count: How STIM and Orai stoichiometry affect store-operated calcium entry. Cell Calcium 2019, 79, 35–43.
- Hoover, P.J.; Lewis, R.S. Stoichiometric requirements for trapping and gating of Ca2+ release-activated Ca2+ (CRAC) channels by stromal interaction molecule 1 (STIM1). Proc. Natl. Acad. Sci. USA 2011, 108, 13299–13304.
- Stathopulos, P.B.; Schindl, R.; Fahrner, M.; Zheng, L.; Gasmi-Seabrook, G.M.; Muik, M.; Romanin, C.; Ikura, M. STIM1/Orai1 coiled-coil interplay in the regulation of store-operated calcium entry. Nat. Commun. 2013, 4, 2963.
- Zhou, Y.; Wang, X.; Wang, X.; Loktionova, N.A.; Cai, X.; Nwokonko, R.M.; Vrana, E.; Wang, Y.; Rothberg, B.S.; Gill, D.L. STIM1 dimers undergo unimolecular coupling to activate Orai1 channels. Nat. Commun. 2015, 6, 8395.
- Zhou, Y.; Nwokonko, R.M.; Cai, X.; Loktionova, N.A.; Abdulqadir, R.; Xin, P.; Niemeyer, B.A.; Wang, Y.; Trebak, M.; Gill, D.L. Cross-linking of Orai1 channels by STIM proteins. Proc. Natl. Acad. Sci. USA 2018, 115, E3398–E3407.
- Shen, Y.; Thillaiappan, N.B.; Taylor, C.W. The store-operated Ca 2+ entry complex comprises a small cluster of STIM1 associated with one Orai1 channel. Proc. Natl. Acad. Sci. USA 2021, 118, e2010789118.
- Rahman, S.; Rahman, T. Unveiling some FDA-approved drugs as inhibitors of the store-operated Ca 2+ entry pathway. Sci. Rep. 2017, 7, 12881.
- Silva-Rojas, R.; Charles, A.L.; Djeddi, S.; Geny, B.; Laporte, J.; Böhm, J. Pathophysiological Effects of Overactive STIM1 on Murine Muscle Function and Structure. Cells 2021, 10, 1730.
- Riva, B.; Griglio, A.; Serafini, M.; Cordero-Sanchez, C.; Aprile, S.; Di Paola, R.; Gugliandolo, E.; Alansary, D.; Biocotino, I.; Lim, D.; et al. Pyrtriazoles, a Novel Class of Store-Operated Calcium Entry Modulators: Discovery, Biological Profiling, and in Vivo Proof-of-Concept Efficacy in Acute Pancreatitis. J. Med. Chem. 2018, 61, 9756–9783.
- Le Guilcher, C.; Luyten, T.; Parys, J.B.; Pucheault, M.; Dellis, O. Synthesis and Characterization of Store-Operated Calcium Entry Inhibitors Active in the Submicromolar Range. Int. J. Mol. Sci. 2020, 21, 9777.
- Meizoso-Huesca, A.; Launikonis, B.S. The Orai1 inhibitor BTP2 has multiple effects on Ca2+ handling in skeletal muscle. J. Gen. Physiol. 2021, 153, e202012747.
- Shimizu-Motohashi, Y.; Komaki, H.; Motohashi, N.; Takeda, S.; Yokota, T.; Aoki, Y. Restoring Dystrophin Expression in Duchenne Muscular Dystrophy: Current Status of Therapeutic Approaches. J. Pers. Med. 2019, 9, 1.
- McMillan, H.J. Intermittent glucocorticoid regimes for younger boys with duchenne muscular dystrophy: Balancing efficacy with side effects. Muscle Nerve 2019, 59, 638–639.
- Bello, L.; Gordish-Dressman, H.; Morgenroth, L.P.; Henricson, E.K.; Duong, T.; Hoffman, E.P.; Cnaan, A.; McDonald, C.M. Prednisone/prednisolone and deflazacort regimens in the CINRG Duchenne Natural History Study. Neurology 2015, 85, 1048–1055.
- Heslop, E.; Turner, C.; Irvin, A.; Muntoni, F.; Straub, V.; Guglieri, M. Gene therapy in Duchenne muscular dystrophy: Identifying and preparing for the challenges ahead. Neuromusc. Dis. 2021, 31, 69–78.
- Fortunato, F.; Rossi, R.; Falzarano, M.S.; Ferlini, A. Innovative Therapeutic Approaches for Duchenne Muscular Dystrophy. J. Clin. Med. 2021, 10, 820.
- Yeung, E.W.; Whitehead, N.P.; Suchyna, T.M.; Gottlieb, P.A.; Sachs, F.; Allen, D.G. Effects of stretch-activated channel blockers on i and muscle damage in the mdx mouse. J. Physiol. 2005, 562 Pt 2, 367–380.
- Cereda, E.; Turri, A.; Klersy, C.; Cappello, S.; Ferrari, A.; Filippi, A.R.; Brugnatelli, S.; Caraccia, M.; Chiellino, S.; Borioli, V.; et al. Whey protein isolate supplementation improves body composition, muscle strength, and treatment tolerance in malnourished advanced cancer patients undergoing chemotherapy. Cancer Med. 2019, 8, 6923–6932.
- Anderson, L.J.; Albrecht, E.D.; Garcia, J.M. Update on management of cancer-related cachexia. Curr. Oncol. Rep. 2017, 19, 3.
- Pierno, S.; De Luca, A.; Desaphy, J.F.; Fraysse, B.; Liantonio, A.; Didonna, M.P.; Lograno, M.; Cocchi, D.; Smith, R.G.; Conte Camerino, D. Growth hormone secretagogues modulate the electrical and contractile properties of rat skeletal muscle through a ghrelin-specific receptor. Br. J. Pharmacol. 2003, 139, 575–584.
- Chen, J.A.; Splenser, A.; Guillory, B.; Luo, J.; Mendiratta, M.; Belinova, B.; Halder, T.; Zhang, G.; Li, Y.P.; Garcia, J.M. Ghrelin prevents tumour- and cisplatin-induced muscle wasting: Characterization of multiple mechanisms involved. J. Cachexia Sarcopenia Muscle 2015, 6, 132–143.
- Von Haehling, S.; Anker, S.D. Treatment of cachexia: An overview of recent developments. Int. J. Cardiol. 2015, 184, 736–742.
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735.

