 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Takeshi Harada | + 6649 word(s) | 6649 | 2021-09-07 05:47:42 | | | |
| 2 | Vivi Li | Meta information modification | 6649 | 2021-10-27 03:18:24 | | | | |
| 3 | Vivi Li | + 155 word(s) | 6804 | 2021-10-27 03:21:17 | | |
Video Upload Options
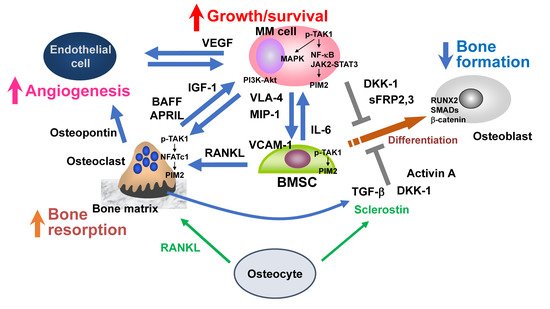
Multiple myeloma (MM) has a propensity to develop preferentially in bone and form bone-destructive lesions. MM cells enhance osteoclastogenesis and bone resorption through activation of the RANKL–NF-κB signaling pathway while suppressing bone formation by inhibiting osteoblastogenesis from bone marrow stromal cells (BMSCs) by factors elaborated in the bone marrow and bone in MM, including the soluble Wnt inhibitors DKK-1 and sclerostin, activin A, and TGF-β, resulting in systemic bone destruction with loss of bone. Osteocytes have been drawn attention as multifunctional regulators in bone metabolism. MM cells induce apoptosis in osteocytes to trigger the production of factors, including RANKL, sclerostin, and DKK-1, to further exacerbate bone destruction. Bone lesions developed in MM, in turn, provide microenvironments suited for MM cell growth/survival, including niches to foster MM cells and their precursors.
1. Introduction
2. Bone Destruction in MM
3. MM Niche
3.1. BMSCs/OBs
3.2. Osteocytes
3.3. OCs
3.4. Vascular Endothelial Cells
3.5. Adipocytes
3.6. MM Tumorigenicity and Plasticity in the Bone Marrow
3.7. The Role of Exosomes and miRNA
3.8. The Effects of MM Bone Microenvironment on Immune Homeostasis
4. Targeting MM Cell–MM Niche Interaction
4.1. Targeting of Osteoclastogenesis and Acidic Microenvironments
| Target Molecules | Agents under Preclinical Investigation | Clinically Available Agents | |
|---|---|---|---|
| MM–OCs interaction | Mevalonate pathway RANK–RANKL CXCR4/SDF-1 MIP-1α IL-3 IL-17 BAFF PIM2, TAK1 |
CXCR4 inhibitor [93] CCR1 inhibitor [94] Anti-IL-3 mAb [95] Anti-IL-17A mAb [96] Anti-BAFF mAb [97] PIM inhibitor [98][99], TAK1 inhibitor [100] |
Bisphosphonate [82] Anti-RANKL mAb [83] |
| MM–BMSC/OB interaction | Sclerostin DKK-1 Notch pathway IL-7 TGF-β Activin A Adiponectin PIM2, TAK1 RUNX2, ATF4 |
Anti-DKK1 mAb [101] γ-secretase inhibitor [102], HDAC inhibitor [103] Anti-IL-7 polyclonal Ab [104] TGF-β receptor inhibitor [17] Recombinant fusion protein of activin receptor type IIA and human IgG1 Fc domain [105] Apolipoprotein A1 mimetic peptide [27] PIM inhibitor [99][106], TAK1 inhibitor [100] |
Anti-sclerostin mAb [107] (available for osteoporosis) Proteasome inhibitors |
4.2. The Role of MM Metabolism in the Bone Marrow
4.3. Induction of Bone Formation
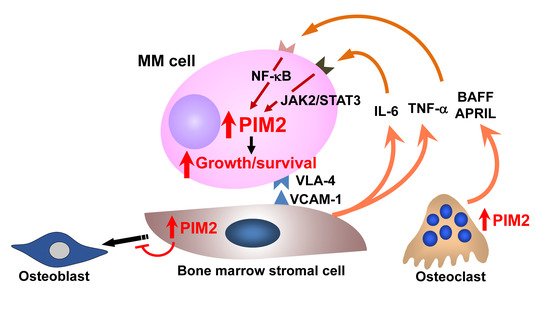
4.4. Targeting PIM Kinases
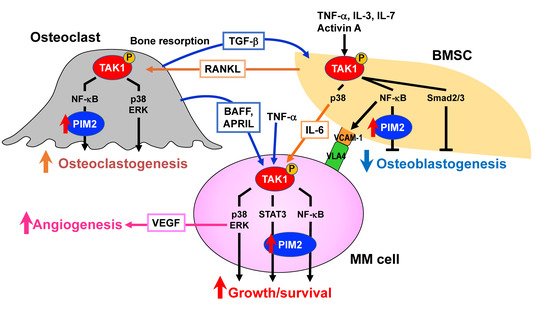
4.5. Targeting TAK1
References
- Lum, L.; Wong, B.R.; Josien, R.; Becherer, J.D.; Erdjument-Bromage, H.; Schlondorff, J.; Tempst, P.; Choi, Y.; Blobel, C.P. Evidence for a Role of a Tumor Necrosis Factor-α (TNF-α)-converting Enzyme-like Protease in Shedding of TRANCE, a TNF Family Member Involved in Osteoclastogenesis and Dendritic Cell Survival. J. Biol. Chem. 1999, 274, 13613–13618.
- Lacey, D.; Timms, E.; Tan, H.-L.; Kelley, M.; Dunstan, C.; Burgess, T.; Elliott, R.; Colombero, A.; Elliott, G.; Scully, S.; et al. Osteoprotegerin Ligand Is a Cytokine that Regulates Osteoclast Differentiation and Activation. Cell 1998, 93, 165–176.
- Zaidi, M. Skeletal remodeling in health and disease. Nat. Med. 2007, 13, 791–801.
- Parfitt, A. Targeted and nontargeted bone remodeling: Relationship to basic multicellular unit origination and progression. Bone 2002, 30, 5–7.
- Nakashima, T.; Hayashi, M.; Takayanagi, H. New insights into osteoclastogenic signaling mechanisms. Trends Endocrinol. Metab. 2012, 23, 582–590.
- O’Brien, C.A.; Nakashima, T.; Takayanagi, H. Osteocyte control of osteoclastogenesis. Bone 2012, 54, 258–263.
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-Hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234.
- Poole, K.E.S.; Van Bezooijen, R.L.; Loveridge, N.; Hamersma, H.; Papapoulos, S.E.; Löwik, C.W.; Reeve, J. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005, 19, 1842–1844.
- Li, J.; Sarosi, I.; Cattley, R.C.; Pretorius, J.; Asuncion, F.; Grisanti, M.; Morony, S.; Adamu, S.; Geng, Z.; Qiu, W.; et al. Dkk1-mediated inhibition of Wnt signaling in bone results in osteopenia. Bone 2006, 39, 754–766.
- Abe, M. Targeting the interplay between myeloma cells and the bone marrow microenvironment in myeloma. Int. J. Hematol. 2011, 94, 334–343.
- Raje, N.; Roodman, G.D. Advances in the Biology and Treatment of Bone Disease in Multiple Myeloma. Clin. Cancer Res. 2011, 17, 1278–1286.
- Choi, S.J.; Cruz, J.C.; Craig, F.; Chung, H.; Devlin, R.D.; Roodman, G.D.; Alsina, M. Macrophage inflammatory protein 1-alpha is a potential osteoclast stimulatory factor in multiple myeloma. Blood 2000, 96, 671–675.
- Han, J.-H.; Choi, S.J.; Kurihara, N.; Koide, M.; Oba, Y.; Roodman, G.D. Macrophage inflammatory protein-1α is an osteoclastogenic factor in myeloma that is independent of receptor activator of nuclear factor κB ligand. Blood 2001, 97, 3349–3353.
- Abe, M.; Hiura, K.; Wilde, J.; Moriyama, K.; Hashimoto, T.; Ozaki, S.; Wakatsuki, S.; Kosaka, M.; Kido, S.; Inoue, D.; et al. Role for macrophage inflammatory protein (MIP)-1alpha and MIP-1beta in the development of osteolytic lesions in multiple myeloma. Blood 2002, 100, 2195–2202.
- Tian, E.; Zhan, F.; Walker, R.; Rasmussen, E.; Ma, Y.; Barlogie, B.; Shaughnessy, J.D. The Role of the Wnt-Signaling Antagonist DKK1 in the Development of Osteolytic Lesions in Multiple Myeloma. N. Engl. J. Med. 2003, 349, 2483–2494.
- Oshima, T.; Abe, M.; Asano, J.; Hara, T.; Kitazoe, K.; Sekimoto, E.; Tanaka, Y.; Shibata, H.; Hashimoto, T.; Ozaki, S.; et al. Myeloma cells suppress bone formation by secreting a soluble Wnt inhibitor, sFRP-2. Blood 2005, 106, 3160–3165.
- Takeuchi, K.; Abe, M.; Hiasa, M.; Oda, A.; Amou, H.; Kido, S.; Harada, T.; Tanaka, O.; Miki, H.; Nakamura, S.; et al. TGF-β Inhibition Restores Terminal Osteoblast Differentiation to Suppress Myeloma Growth. PLoS ONE 2010, 5, e9870.
- Yaccoby, S.; Pearse, R.N.; Johnson, C.L.; Barlogie, B.; Choi, Y.; Epstein, J. Myeloma interacts with the bone marrow microenvironment to induce osteoclastogenesis and is dependent on osteoclast activity. Br. J. Haematol. 2002, 116, 278–290.
- Liu, J.; Hamrouni, A.; Wolowiec, D.; Coiteux, V.; Kuliczkowski, K.; Hetuin, D.; Saudemont, A.; Quesnel, B. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-γ and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood 2007, 110, 296–304.
- An, G.; Acharya, C.; Feng, X.; Wen, K.; Zhong, M.; Zhang, L.; Munshi, N.C.; Qiu, L.; Tai, Y.-T.; Anderson, K.C. Osteoclasts promote immune suppressive microenvironment in multiple myeloma: Therapeutic implication. Blood 2016, 128, 1590–1603.
- Iwasa, M.; Harada, T.; Oda, A.; Bat-Erdene, A.; Teramachi, J.; Tenshin, H.; Ashtar, M.; Oura, M.; Sogabe, K.; Udaka, K.; et al. PD-L1 upregulation in myeloma cells by panobinostat in combination with interferon-γ. Oncotarget 2019, 10, 1903–1917.
- Ray, A.; Das, D.S.; Song, Y.; Richardson, P.; Munshi, N.C.; Chauhan, D.; Anderson, K.C. Targeting PD1–PDL1 immune checkpoint in plasmacytoid dendritic cell interactions with T cells, natural killer cells and multiple myeloma cells. Leukemia 2015, 29, 1441–1444.
- Görgün, G.T.; Samur, M.K.; Cowens, K.B.; Paula, S.; Bianchi, G.; Anderson, J.E.; White, R.E.; Singh, A.; Ohguchi, H.; Suzuki, R.; et al. Lenalidomide Enhances Immune Checkpoint Blockade-Induced Immune Response in Multiple Myeloma. Clin. Cancer Res. 2015, 21, 4607–4618.
- Landowski, T.H.; E Olashaw, N.; Agrawal, D.; Dalton, W.S. Cell adhesion-mediated drug resistance (CAM-DR) is associated with activation of NF-κB (RelB/p50) in myeloma cells. Oncogene 2003, 22, 2417–2421.
- Mori, Y.; Shimizu, N.; Dallas, M.; Niewolna, M.; Story, B.; Williams, P.J.; Mundy, G.R.; Yoneda, T.; Harris, M.N.; Ozpolat, B.; et al. Anti-α4 integrin antibody suppresses the development of multiple myeloma and associated osteoclastic osteolysis. Blood 2004, 104, 2149–2154.
- Abe, M.; Hiura, K.; Ozaki, S.; Kido, S.; Matsumoto, T. Vicious cycle between myeloma cell binding to bone marrow stromal cells via VLA-4–VCAM-1 adhesion and macrophage inflammatory protein-1α and MIP-1β production. J. Bone Miner. Metab. 2008, 27, 16–23.
- Fowler, J.A.; Lwin, S.T.; Drake, M.T.; Edwards, J.R.; Kyle, R.A.; Mundy, G.R.; Edwards, C. Host-derived adiponectin is tumor-suppressive and a novel therapeutic target for multiple myeloma and the associated bone disease. Blood 2011, 118, 5872–5882.
- Chen, Z.; Orlowski, R.; Wang, M.; Kwak, L.; Mccarty, N. Osteoblastic niche supports the growth of quiescent multiple myeloma cells. Blood 2014, 123, 2204–2208.
- Lawson, M.A.; McDonald, M.; Kovacic, N.; Khoo, W.H.; Terry, R.L.; Down, J.M.; Kaplan, W.; Paton-Hough, J.; Fellows, C.; Pettitt, J.A.; et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat. Commun. 2015, 6, 8983.
- Bonewald, L.F. The amazing osteocyte. J. Bone Miner. Res. 2011, 26, 229–238.
- Xiong, J.; Onal, M.; Jilka, R.L.; Weinstein, R.S.; Manolagas, S.C.; O’Brien, C.A. Matrix-embedded cells control osteoclast formation. Nat. Med. 2011, 17, 1235–1241.
- Kramer, I.; Halleux, C.; Keller, H.; Pegurri, M.; Gooi, J.; Weber, P.B.; Feng, J.Q.; Bonewald, L.F.; Kneissel, M. Osteocyte Wnt/β-Catenin Signaling Is Required for Normal Bone Homeostasis. Mol. Cell. Biol. 2010, 30, 3071–3085.
- Delgado-Calle, J.; Anderson, J.; Cregor, M.D.; Hiasa, M.; Chirgwin, J.M.; Carlesso, N.; Yoneda, T.; Mohammad, K.S.; Plotkin, L.I.; Roodman, G.D.; et al. Bidirectional Notch Signaling and Osteocyte-Derived Factors in the Bone Marrow Microenvironment Promote Tumor Cell Proliferation and Bone Destruction in Multiple Myeloma. Cancer Res. 2016, 76, 1089–1100.
- Giuliani, N.; Ferretti, M.; Bolzoni, M.; Storti, P.; Lazzaretti, M.; Palma, A.B.D.; Bonomini, S.; Martella, E.; Agnelli, L.; Neri, A.; et al. Increased osteocyte death in multiple myeloma patients: Role in myeloma-induced osteoclast formation. Leukemia 2012, 26, 1391–1401.
- Toscani, D.; Palumbo, C.; Palma, A.B.D.; Ferretti, M.; Bolzoni, M.; Marchica, V.; Sena, P.; Martella, E.; Mancini, C.; Ferri, V.; et al. The Proteasome Inhibitor Bortezomib Maintains Osteocyte Viability in Multiple Myeloma Patients by Reducing Both Apoptosis and Autophagy: A New Function for Proteasome Inhibitors. J. Bone Miner. Res. 2015, 31, 815–827.
- Robinson, M.K.; Caminis, J.; Brunkow, M.E. Sclerostin: How human mutations have helped reveal a new target for the treatment of osteoporosis. Drug Discov. Today 2013, 18, 637–643.
- Lewiecki, E.M. Role of sclerostin in bone and cartilage and its potential as a therapeutic target in bone diseases. Ther. Adv. Musculoskelet. Dis. 2013, 6, 48–57.
- Delgado-Calle, J.; Anderson, J.; Cregor, M.D.; Condon, K.W.; Kuhstoss, S.A.; Plotkin, L.I.; Bellido, T.; Roodman, G.D. Genetic deletion of Sost or pharmacological inhibition of sclerostin prevent multiple myeloma-induced bone disease without affecting tumor growth. Leukemia 2017, 31, 2686–2694.
- Terpos, E.; Christoulas, D.; Katodritou, E.; Bratengeier, C.; Gkotzamanidou, M.; Michalis, E.; Delimpasi, S.; Pouli, A.; Meletis, J.; Kastritis, E.; et al. Elevated circulating sclerostin correlates with advanced disease features and abnormal bone remodeling in symptomatic myeloma: Reduction post-bortezomib monotherapy. Int. J. Cancer 2011, 131, 1466–1471.
- Loredana, S.; Santo, L.; Wein, M.N.; Hu, D.Z.; Cirstea, D.D.; Nemani, N.; Tai, Y.-T.; Raines, S.E.; Kuhstoss, S.A.; Munshi, N.C.; et al. Regulation of Sclerostin Expression in Multiple Myeloma by Dkk-1: A Potential Therapeutic Strategy for Myeloma Bone Disease. J. Bone Miner. Res. 2016, 31, 1225–1234.
- McDonald, M.M.; Reagan, M.R.; Youlten, S.E.; Mohanty, S.T.; Seckinger, A.; Terry, R.L.; Pettitt, J.A.; Simic, M.K.; Cheng, T.L.; Morse, A.; et al. Inhibiting the osteocyte-specific protein sclerostin increases bone mass and fracture resistance in multiple myeloma. Blood 2017, 129, 3452–3464.
- Mulcrone, P.L.; Edwards, S.K.E.; Petrusca, D.N.; Haneline, L.S.; Delgado-Calle, J.; Roodman, G.D. Osteocyte Vegf-a contributes to myeloma-associated angiogenesis and is regulated by Fgf23. Sci. Rep. 2020, 10, 17319.
- Yaccoby, S.; Wezeman, M.J.; Henderson, A.; Cottler-Fox, M.; Yi, Q.; Barlogie, B.; Epstein, J. Cancer and the Microenvironment: Myeloma-osteoclast interactions as a model. Cancer Res. 2004, 64, 2016–2023.
- Abe, M.; Hiura, K.; Wilde, J.; Shioyasono, A.; Moriyama, K.; Hashimoto, T.; Kido, S.; Oshima, T.; Shibata, H.; Ozaki, S.; et al. Osteoclasts enhance myeloma cell growth and survival via cell-cell contact: A vicious cycle between bone destruction and myeloma expansion. Blood 2004, 104, 2484–2491.
- Novak, A.J.; Darce, J.R.; Arendt, B.K.; Harder, B.; Henderson, K.; Kindsvogel, W.; Gross, J.A.; Greipp, P.R.; Jelinek, D.F. Expression of BCMA, TACI, and BAFF-R in multiple myeloma: A mechanism for growth and survival. Blood 2004, 103, 689–694.
- Moreaux, J.; Legouffe, E.; Jourdan, E.; Quittet, P.; Rème, T.; Lugagne, C.; Moine, P.; Rossi, J.-F.; Klein, B.; Tarte, K. BAFF and APRIL protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood 2004, 103, 3148–3157.
- Moreaux, J.; Cremer, F.W.; Reme, T.; Raab, M.; Mahtouk, K.; Kaukel, P.; Pantesco, V.; DE Vos, J.; Jourdan, E.; Jauch, A.; et al. The level of TACI gene expression in myeloma cells is associated with a signature of microenvironment dependence versus a plasmablastic signature. Blood 2005, 106, 1021–1030.
- Abe, M.; Kido, S.; Hiasa, M.; Nakano, A.; Oda, A.; Amou, H.; Matsumoto, T. BAFF and APRIL as osteoclast-derived survival factors for myeloma cells: A rationale for TACI-Fc treatment in patients with multiple myeloma. Leukemia 2006, 20, 1313–1315.
- Yaccoby, S.; Pennisi, A.; Li, X.; Dillon, S.R.; Zhan, F.; Barlogie, B.; Shaughnessy, J.D., Jr. Atacicept (TACI-Ig) inhibits growth of TACIhigh primary myeloma cells in SCID-hu mice and in coculture with osteoclasts. Leukemia 2007, 22, 406–413.
- Mathupala, S.P.; Rempel, A.; Pedersen, P.L. Glucose catabolism in cancer cells. Isolation, sequence, and activity of the promoter for type ii hexokinase. J. Biol. Chem. 1995, 270, 16918–16925.
- Nakano, A.; Miki, H.; Nakamura, S.; Harada, T.; Oda, A.; Amou, H.; Fujii, S.; Kagawa, K.; Takeuchi, K.; Ozaki, S.; et al. Up-regulation of hexokinaseII in myeloma cells: Targeting myeloma cells with 3-bromopyruvate. J. Bioenerg. Biomembr. 2012, 44, 31–38.
- Nakano, A.; Tsuji, D.; Miki, H.; Cui, Q.; El Sayed, S.M.; Ikegame, A.; Oda, A.; Amou, H.; Nakamura, S.; Harada, T.; et al. Glycolysis Inhibition Inactivates ABC Transporters to Restore Drug Sensitivity in Malignant Cells. PLoS ONE 2011, 6, e27222.
- Pastorino, J.G.; Shulga, N.; Hoek, J. Mitochondrial Binding of Hexokinase II Inhibits Bax-induced Cytochrome c Release and Apoptosis. J. Biol. Chem. 2002, 277, 7610–7618.
- Bhatti, S.S.; Kumar, L.; Dinda, A.K.; Dawar, R. Prognostic value of bone marrow angiogenesis in multiple myeloma: Use of light microscopy as well as computerized image analyzer in the assessment of microvessel density and total vascular area in multiple myeloma and its correlation with various clinical, histological, and laboratory parameters. Am. J. Hematol. 2006, 81, 649–656.
- Jakob, C.; Sterz, J.; Zavrski, I.; Heider, U.; Kleeberg, L.; Fleissner, C.; Kaiser, M.; Sezer, O. Angiogenesis in multiple myeloma. Eur. J. Cancer 2006, 42, 1581–1590.
- Corre, J.; Mahtouk, K.; Attal, M.; Gadelorge, M.; Huynh, A.; Fleury-Cappellesso, S.; Danho, C.; Laharrague, P.; Klein, B.; Rème, T.; et al. Bone marrow mesenchymal stem cells are abnormal in multiple myeloma. Leukemia 2007, 21, 1079–1088.
- Tanaka, Y.; Abe, M.; Hiasa, M.; Oda, A.; Amou, H.; Nakano, A.; Takeuchi, K.; Kitazoe, K.; Kido, S.; Inoue, D.; et al. Myeloma Cell-Osteoclast Interaction Enhances Angiogenesis Together with Bone Resorption: A Role for Vascular Endothelial Cell Growth Factor and Osteopontin. Clin. Cancer Res. 2007, 13, 816–823.
- A Takafuji, V.; Forgues, M.; Unsworth, E.; Goldsmith, P.; Wang, X.W. An osteopontin fragment is essential for tumor cell invasion in hepatocellular carcinoma. Oncogene 2007, 26, 6361–6371.
- Cackowski, F.C.; Anderson, J.L.; Patrene, K.D.; Choksi, R.J.; Shapiro, S.D.; Windle, J.; Blair, H.C.; Roodman, G.D. Osteoclasts are important for bone angiogenesis. Blood 2010, 115, 140–149.
- Liu, Z.; Liu, H.; He, J.; Lin, P.; Tong, Q.; Yang, J. Myeloma cells shift osteoblastogenesis to adipogenesis by inhibiting the ubiquitin ligase MURF1 in mesenchymal stem cells. Sci. Signal. 2020, 13, eaay8203.
- Morris, E.V.; Suchacki, K.J.; Hocking, J.; Cartwright, R.; Sowman, A.; Gamez, B.; Lea, R.; Drake, M.T.; Cawthorn, W.P.; Edwards, C.M.; et al. Myeloma Cells Down-Regulate Adiponectin in Bone Marrow Adipocytes Via TNF-Alpha. J. Bone Miner. Res. 2019, 35, 942–955.
- Fairfield, H.; Dudakovic, A.; Khatib, C.M.; Farrell, M.; Costa, S.; Falank, C.; Hinge, M.; Murphy, C.S.; DeMambro, V.; Pettitt, J.A.; et al. Myeloma-Modified Adipocytes Exhibit Metabolic Dysfunction and a Senescence-Associated Secretory Phenotype. Cancer Res. 2020, 81, 634–647.
- Liu, H.; He, J.; Koh, S.P.; Zhong, Y.; Liu, Z.; Wang, Z.; Zhang, Y.; Li, Z.; Tam, B.T.; Lin, P.; et al. Reprogrammed marrow adipocytes contribute to myeloma-induced bone disease. Sci. Transl. Med. 2019, 11, eaau9087.
- Li, Z.; Liu, H.; He, J.; Wang, Z.; Yin, Z.; You, G.; Wang, Z.; Davis, R.E.; Lin, P.; Bergsagel, P.L.; et al. Acetyl-CoA Synthetase 2: A Critical Linkage in Obesity-Induced Tumorigenesis in Myeloma. Cell Metab. 2021, 33, 78–93.e7.
- Yaccoby, S.; Epstein, J. The Proliferative Potential of Myeloma Plasma Cells Manifest in the SCID-hu Host. Blood 1999, 94, 3576–3582.
- Hosen, N.; Matsuoka, Y.; Kishida, S.; Nakata, J.; Mizutani, Y.; Hasegawa, K.; Mugitani, A.; Ichihara, H.; Aoyama, Y.; Nishida, S.; et al. CD138-negative clonogenic cells are plasma cells but not B cells in some multiple myeloma patients. Leukemia 2012, 26, 2135–2141.
- Yata, K.; Yaccoby, S. The SCID-rab model: A novel in vivo system for primary human myeloma demonstrating growth of CD138-expressing malignant cells. Leukemia 2004, 18, 1891–1897.
- Yaccoby, S. The Phenotypic Plasticity of Myeloma Plasma Cells as Expressed by Dedifferentiation into an Immature, Resilient, and Apoptosis-Resistant Phenotype. Clin. Cancer Res. 2005, 11, 7599–7606.
- Chaidos, A.; Barnes, C.; Cowan, G.; May, P.; Melo, V.; Hatjiharissi, E.; Papaioannou, M.; Harrington, H.; Doolittle, H.; Terpos, E.; et al. Clinical drug resistance linked to interconvertible phenotypic and functional states of tumor-propagating cells in multiple myeloma. Blood 2013, 121, 318–328.
- Li, B.; Xu, H.; Han, H.; Song, S.; Zhang, X.; Ouyang, L.; Qian, C.; Hong, Y.; Qiu, Y.; Zhou, W.; et al. Exosome-mediated transfer of lncRUNX2-AS1 from multiple myeloma cells to MSCs contributes to osteogenesis. Oncogene 2018, 37, 5508–5519.
- Raimondo, S.; Saieva, L.; Vicario, E.; Pucci, M.; Toscani, D.; Manno, M.; Raccosta, S.; Giuliani, N.; Alessandro, R.; Raccosta, S. Multiple myeloma-derived exosomes are enriched of amphiregulin (AREG) and activate the epidermal growth factor pathway in the bone microenvironment leading to osteoclastogenesis. J. Hematol. Oncol. 2019, 12, 2.
- Wang, J.; Hendrix, A.; Hernot, S.; Lemaire, M.; De Bruyne, E.; Van Valckenborgh, E.; Lahoutte, T.; De Wever, O.; Vanderkerken, K.; Menu, E. Bone marrow stromal cell–derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood 2014, 124, 555–566.
- Wang, J.; De Veirman, K.; Faict, S.; Frassanito, M.A.; Ribatti, D.; Vacca, A.; Menu, E. Multiple myeloma exosomes establish a favourable bone marrow microenvironment with enhanced angiogenesis and immunosuppression. J. Pathol. 2016, 239, 162–173.
- Pitari, M.R.; Rossi, M.; Amodio, N.; Botta, C.; Morelli, E.; Federico, C.; Gullà, A.; Caracciolo, D.; Di Martino, M.T.; Arbitrio, M.; et al. Inhibition of miR-21 restores RANKL/OPG ratio in multiple myeloma-derived bone marrow stromal cells and impairs the resorbing activity of mature osteoclasts. Oncotarget 2015, 6, 27343–27358.
- Gowda, P.S.; Wildman, B.J.; Trotter, T.N.; Xu, X.; Hao, X.; Hassan, M.Q.; Yang, Y. Runx2 Suppression by miR-342 and miR-363 Inhibits Multiple Myeloma Progression. Mol. Cancer Res. 2018, 16, 1138–1148.
- Fan, F.; Deng, R.; Qiu, L.; Wen, Q.; Zeng, Y.; Gao, L.; Zhang, C.; Kong, P.; Zhong, J.; Zeng, N.; et al. miR-203a-3p.1 is involved in the regulation of osteogenic differentiation by directly targeting Smad9 in MM-MSCs. Oncol. Lett. 2019, 18, 6339–6346.
- Papanota, A.-M.; Karousi, P.; Kontos, C.; Ntanasis-Stathopoulos, I.; Scorilas, A.; Terpos, E. Multiple Myeloma Bone Disease: Implication of MicroRNAs in Its Molecular Background. Int. J. Mol. Sci. 2021, 22, 2375.
- Harada, T.; Miki, H.; Cui, Q.; Oda, A.; Amachi, R.; Teramachi, J.; Bat-Erdene, A.; Sogabe, K.; Iwasa, M.; Fujii, S.; et al. Expansion of Th1-like Vγ9Vδ2T cells by new-generation IMiDs, lenalidomide and pomalidomide, in combination with zoledronic acid. Leukemia 2016, 31, 258–262.
- Castella, B.; Foglietta, M.; Riganti, C.; Massaia, M. Vγ9Vδ2 T Cells in the Bone Marrow of Myeloma Patients: A Paradigm of Microenvironment-Induced Immune Suppression. Front. Immunol. 2018, 9, 1492.
- Croucher, P.; Shipman, C.M.; Lippitt, J.; Perry, M.; Asosingh, K.; Hijzen, A.; Brabbs, A.C.; Van Beek, E.J.R.; Holen, I.; Skerry, T.; et al. Osteoprotegerin inhibits the development of osteolytic bone disease in multiple myeloma. Blood 2001, 98, 3534–3540.
- I Croucher, P.; De Raeve, H.; Perry, M.J.; Hijzen, A.; Shipman, C.M.; Lippitt, J.; Green, J.; Van Marck, E.; Van Camp, B.; Vanderkerken, K. Zoledronic Acid Treatment of 5T2MM-Bearing Mice Inhibits the Development of Myeloma Bone Disease: Evidence for Decreased Osteolysis, Tumor Burden and Angiogenesis, and Increased Survival. J. Bone Miner. Res. 2003, 18, 482–492.
- Morgan, G.J.; Davies, F.; Gregory, W.M.; Cocks, K.; E Bell, S.; Szubert, A.J.; Navarro-Coy, N.; Drayson, M.; Owen, R.G.; Feyler, S.; et al. First-line treatment with zoledronic acid as compared with clodronic acid in multiple myeloma (MRC Myeloma IX): A randomised controlled trial. Lancet 2010, 376, 1989–1999.
- Raje, N.; Terpos, E.; Willenbacher, W.; Shimizu, K.; Garcia-Sanz, R.; Durie, B.; Legieć, W.; Krejčí, M.; Laribi, K.; Zhu, L.; et al. Denosumab versus zoledronic acid in bone disease treatment of newly diagnosed multiple myeloma: An international, double-blind, double-dummy, randomised, controlled, phase 3 study. Lancet Oncol. 2018, 19, 370–381.
- Ji, K.; Mayernik, L.; Moin, K.; Sloane, B.F. Acidosis and proteolysis in the tumor microenvironment. Cancer Metastasis Rev. 2019, 38, 103–112.
- Teitelbaum, S. Bone Resorption by Osteoclasts. Science 2000, 289, 1504–1508.
- Amachi, R.; Hiasa, M.; Teramachi, J.; Harada, T.; Oda, A.; Nakamura, S.; Hanson, D.; Watanabe, K.; Fujii, S.; Miki, H.; et al. A vicious cycle between acid sensing and survival signaling in myeloma cells: Acid-induced epigenetic alteration. Oncotarget 2016, 7, 70447–70461.
- Gerweck, L.E.; Vijayappa, S.; Kozin, S. Tumor pH controls the in vivo efficacy of weak acid and base chemotherapeutics. Mol. Cancer Ther. 2006, 5, 1275–1279.
- Tannock, I.F.; Rotin, D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 1989, 49, 4373–4384.
- Kawatani, M.; Osada, H. Osteoclast-targeting small molecules for the treatment of neoplastic bone metastases. Cancer Sci. 2009, 100, 1999–2005.
- Woo, J.-T.; Kawatani, M.; Kato, M.; Shinki, T.; Yonezawa, T.; Kanoh, N.; Nakagawa, H.; Takami, M.; Lee, K.H.; Stern, P.H.; et al. Reveromycin A, an agent for osteoporosis, inhibits bone resorption by inducing apoptosis specifically in osteoclasts. Proc. Natl. Acad. Sci. USA 2006, 103, 4729–4734.
- Muguruma, H.; Yano, S.; Kakiuchi, S.; Uehara, H.; Kawatani, M.; Osada, H.; Sone, S. Reveromycin A Inhibits Osteolytic Bone Metastasis of Small-Cell Lung Cancer Cells, SBC-5, through an Antiosteoclastic Activity. Clin. Cancer Res. 2005, 11, 8822–8828.
- Watanabe, K.; Bat-Erdene, A.; Tenshin, H.; Cui, Q.; Teramachi, J.; Hiasa, M.; Oda, A.; Harada, T.; Miki, H.; Sogabe, K.; et al. Reveromycin A, a novel acid-seeking agent, ameliorates bone destruction and tumor growth in multiple myeloma. Haematologica 2020, 106, 1172–1177.
- Zannettino, A.C.; Farrugia, A.N.; Kortesidis, A.; Manavis, J.; To, L.B.; Martin, S.; Diamond, P.; Tamamura, H.; Lapidot, T.; Fujii, N.; et al. Elevated Serum Levels of Stromal-Derived Factor-1α Are Associated with Increased Osteoclast Activity and Osteolytic Bone Disease in Multiple Myeloma Patients. Cancer Res. 2005, 65, 1700–1709.
- Vallet, S.; Raje, N.; Ishitsuka, K.; Hideshima, T.; Podar, K.; Chhetri, S.; Pozzi, S.; Breitkreutz, I.; Kiziltepe, T.; Yasui, H.; et al. MLN3897, a novel CCR1 inhibitor, impairs osteoclastogenesis and inhibits the interaction of multiple myeloma cells and osteoclasts. Blood 2007, 110, 3744–3752.
- Lee, J.W.; Chung, H.Y.; Ehrlich, L.A.; Jelinek, D.F.; Callander, N.S.; Roodman, G.D.; Choi, S.J. IL-3 expression by myeloma cells increases both osteoclast formation and growth of myeloma cells. Blood 2004, 103, 2308–2315.
- Prabhala, R.H.; Fulciniti, M.; Pelluru, D.; Rashid, N.U.; Nigroiu, A.; Nanjappa, P.; Pai, C.; Lee, S.; Prabhala, N.S.; Bandi, R.L.; et al. Targeting IL-17A in multiple myeloma: A potential novel therapeutic approach in myeloma. Leukemia 2015, 30, 379–389.
- Raje, N.S.; Moreau, P.; Terpos, E.; Benboubker, L.; Grząśko, N.; Holstein, S.A.; Oriol, A.; Huang, S.; Beksac, M.; Kuliczkowski, K.; et al. Phase 2 study of tabalumab, a human anti-B-cell activating factor antibody, with bortezomib and dexamethasone in patients with previously treated multiple myeloma. Br. J. Haematol. 2016, 176, 783–795.
- Teramachi, J.; Hiasa, M.; Oda, A.; Harada, T.; Nakamura, S.; Amachi, R.; Tenshin, H.; Iwasa, M.; Fujii, S.; Kagawa, K.; et al. Pim-2 is a critical target for treatment of osteoclastogenesis enhanced in myeloma. Br. J. Haematol. 2016, 180, 581–585.
- Paíno, T.; Garcia-Gomez, A.; González-Méndez, L.; San-Segundo, L.; Hernández-García, S.; López-Iglesias, A.-A.; Algarín, E.M.; Martín-Sánchez, M.; Corbacho-González, D.; Ortiz-De-Solorzano, C.; et al. The Novel Pan-PIM Kinase Inhibitor, PIM447, Displays Dual Antimyeloma and Bone-Protective Effects, and Potently Synergizes with Current Standards of Care. Clin. Cancer Res. 2016, 23, 225–238.
- Teramachi, J.; Tenshin, H.; Hiasa, M.; Oda, A.; Bat-Erdene, A.; Harada, T.; Nakamura, S.; Ashtar, M.; Shimizu, S.; Iwasa, M.; et al. TAK1 is a pivotal therapeutic target for tumor progression and bone destruction in myeloma. Haematologica 2020, 106, 1401–1413.
- Fulciniti, M.; Tassone, P.; Hideshima, T.; Vallet, S.; Nanjappa, P.; Ettenberg, S.A.; Shen, Z.; Patel, N.; Tai, Y.-T.; Chauhan, D.; et al. Anti-DKK1 mAb (BHQ880) as a potential therapeutic agent for multiple myeloma. Blood 2009, 114, 371–379.
- Xu, S.; Evans, H.; Buckle, C.; De Veirman, K.; Hu, J.; Xu, D.; Menu, E.; De Becker, A.; Broek, I.V.; Leleu, X.; et al. Impaired osteogenic differentiation of mesenchymal stem cells derived from multiple myeloma patients is associated with a blockade in the deactivation of the Notch signaling pathway. Leukemia 2012, 26, 2546–2549.
- Adamik, J.; Jin, S.; Sun, Q.; Zhang, P.; Weiss, K.R.; Anderson, J.L.; Silbermann, R.; Roodman, G.D.; Galson, D.L. EZH2 or HDAC1 Inhibition Reverses Multiple Myeloma–Induced Epigenetic Suppression of Osteoblast Differentiation. Mol. Cancer Res. 2017, 15, 405–417.
- Giuliani, N.; Colla, S.; Morandi, F.; Lazzaretti, M.; Sala, R.; Bonomini, S.; Grano, M.; Colucci, S.; Svaldi, M.; Rizzoli, V. Myeloma cells block RUNX2/CBFA1 activity in human bone marrow osteoblast progenitors and inhibit osteoblast formation and differentiation. Blood 2005, 106, 2472–2483.
- Abdulkadyrov, K.M.; Salogub, G.N.; Khuazheva, N.K.; Sherman, M.L.; Laadem, A.; Barger, R.; Knight, R.; Srinivasan, S.; Terpos, E. Sotatercept in patients with osteolytic lesions of multiple myeloma. Br. J. Haematol. 2014, 165, 814–823.
- Hiasa, M.; Teramachi, J.; Oda, A.; Amachi, R.; Harada, T.; Nakamura, S.; Miki, H.; Fujii, S.; Kagawa, K.; Watanabe, K.; et al. Pim-2 kinase is an important target of treatment for tumor progression and bone loss in myeloma. Leukemia 2014, 29, 207–217.
- Munshi, N.C.; Abonour, R.; Beck, J.T.; Bensinger, W.; Facon, T.; Stockerl-Goldstein, K.; Baz, R.; Siegel, D.S.; Neben, K.; Lonial, S.; et al. Early Evidence of Anabolic Bone Activity of BHQ880, a Fully Human Anti-DKK1 Neutralizing Antibody: Results of a Phase 2 Study in Previously Untreated Patients with Smoldering Multiple Myeloma At Risk for Progression. Blood 2012, 120, 331.
- Altman, B.; Stine, Z.E.; Dang, C. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634.
- Puchades-Carrasco, L.; Lecumberri, R.; Martinez-Lopez, J.; Lahuerta, J.-J.; Mateos, M.-V.; Prosper, F.; Miguel, J.S.; Pineda-Lucena, A. Multiple Myeloma Patients Have a Specific Serum Metabolomic Profile That Changes after Achieving Complete Remission. Clin. Cancer Res. 2013, 19, 4770–4779.
- Bolzoni, M.; Chiu, M.; Accardi, F.; Vescovini, R.; Airoldi, I.; Storti, P.; Todoerti, K.; Agnelli, L.; Missale, G.; Andreoli, R.; et al. Dependence on glutamine uptake and glutamine addiction characterize myeloma cells: A new attractive target. Blood 2016, 128, 667–679.
- He, Y.; Wang, Y.; Liu, H.; Xu, X.; He, S.; Tang, J.; Huang, Y.; Miao, X.; Wu, Y.; Wang, Q.; et al. Pyruvate kinase isoform M2 (PKM2) participates in multiple myeloma cell proliferation, adhesion and chemoresistance. Leuk. Res. 2015, 39, 1428–1436.
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370.
- Beharry, Z.; Mahajan, S.; Zemskova, M.; Lin, Y.-W.; Tholanikunnel, B.G.; Xia, Z.; Smith, C.D.; Kraft, A.S. The Pim protein kinases regulate energy metabolism and cell growth. Proc. Natl. Acad. Sci. USA 2010, 108, 528–533.
- Zhang, H.; Li, L.; Chen, Q.; Li, M.; Feng, J.; Sun, Y.; Zhao, R.; Zhu, Y.; Lv, Y.; Zhu, Z.; et al. PGC 1β regulates multiple myeloma tumor growth through LDHA -mediated glycolytic metabolism. Mol. Oncol. 2018, 12, 1579–1595.
- Lund, T.; Søe, K.; Abildgaard, N.; Garnero, P.; Pedersen, P.T.; Ormstrup, T.; Delaisse, J.-M.; Plesner, T. First-line treatment with bortezomib rapidly stimulates both osteoblast activity and bone matrix deposition in patients with multiple myeloma, and stimulates osteoblast proliferation and differentiation in vitro. Eur. J. Haematol. 2010, 85, 290–299.
- Lee, S.-E.; Min, C.-K.; Yahng, S.-A.; Cho, B.-S.; Eom, K.-S.; Kim, Y.-J.; Kim, H.-J.; Lee, S.; Cho, S.-G.; Kim, N.-W.; et al. Bone scan images reveal increased osteoblastic function after bortezomib treatment in patients with multiple myeloma. Eur. J. Haematol. 2010, 86, 83–86.
- Delforge, M.; Terpos, E.; Richardson, P.G.; Shpilberg, O.; Khuageva, N.K.; Schlag, R.; Dimopoulos, M.; Kropff, M.; Spicka, I.; Petrucci, M.T.; et al. Fewer bone disease events, improvement in bone remodeling, and evidence of bone healing with bortezomib plus melphalan-prednisone vs. melphalan-prednisone in the phase III VISTA trial in multiple myeloma. Eur. J. Haematol. 2011, 86, 372–384.
- Yaccoby, S.; Wezeman, M.J.; Zangari, M.; Walker, R.; Cottler-Fox, M.; Gaddy, D.; Ling, W.; Saha, R.; Barlogie, B.; Tricot, G.; et al. Inhibitory effects of osteoblasts and increased bone formation on myeloma in novel culture systems and a myelomatous mouse model. Haematologica 2006, 91, 192–199.
- Yaccoby, S.; Ling, W.; Zhan, F.; Walker, R.; Barlogie, B.; Shaughnessy, J.D. Antibody-based inhibition of DKK1 suppresses tumor-induced bone resorption and multiple myeloma growth in vivo. Blood 2006, 109, 2106–2111.
- Heath, D.J.; Chantry, A.D.; Buckle, C.H.; Coulton, L.; Shaughnessy, J.D.; Evans, H.R.; A Snowden, J.; Stover, D.R.; Vanderkerken, K.; I Croucher, P. Inhibiting Dickkopf-1 (Dkk1) Removes Suppression of Bone Formation and Prevents the Development of Osteolytic Bone Disease in Multiple Myeloma. J. Bone Miner. Res. 2009, 24, 425–436.
- Edwards, C.M.; Edwards, J.R.; Lwin, S.T.; Esparza, J.; Oyajobi, B.O.; McCluskey, B.; Munoz, S.; Grubbs, B.; Mundy, G.R. Increasing Wnt signaling in the bone marrow microenvironment inhibits the development of myeloma bone disease and reduces tumor burden in bone in vivo. Blood 2008, 111, 2833–2842.
- Vallet, S.; Mukherjee, S.; Vaghela, N.; Hideshima, T.; Fulciniti, M.; Pozzi, S.; Santo, L.; Cirstea, D.; Patel, K.; Sohani, A.R.; et al. Activin A promotes multiple myeloma-induced osteolysis and is a promising target for myeloma bone disease. Proc. Natl. Acad. Sci. USA 2010, 107, 5124–5129.
- Chantry, A.; Heath, D.; Mulivor, A.W.; Pearsall, S.; Baud’Huin, M.; Coulton, L.; Evans, H.; Abdul, N.; Werner, E.D.; Bouxsein, M.L.; et al. Inhibiting activin-A signaling stimulates bone formation and prevents cancer-induced bone destruction in vivo. J. Bone Miner. Res. 2010, 25, 2633–2646.
- Qiang, Y.-W.; Shaughnessy, J.D.; Yaccoby, S. Wnt3a signaling within bone inhibits multiple myeloma bone disease and tumor growth. Blood 2008, 112, 374–382.
- Maeda, S.; Hayashi, M.; Komiya, S.; Imamura, T.; Miyazono, K. Endogenous TGF-β signaling suppresses maturation of osteoblastic mesenchymal cells. EMBO J. 2004, 23, 552–563.
- Matsumoto, T.; Abe, M. TGF-β-related mechanisms of bone destruction in multiple myeloma. Bone 2011, 48, 129–134.
- Lotinun, S.; Pearsall, R.S.; Davies, M.V.; Marvell, T.H.; Monnell, T.E.; Ucran, J.; Fajardo, R.J.; Kumar, R.; Underwood, K.W.; Seehra, J.; et al. A soluble activin receptor Type IIA fusion protein (ACE-011) increases bone mass via a dual anabolic-antiresorptive effect in Cynomolgus monkeys. Bone 2010, 46, 1082–1088.
- Asano, J.; Nakano, A.; Oda, A.; Amou, H.; Hiasa, M.; Takeuchi, K.; Miki, H.; Nakamura, S.; Harada, T.; Fujii, S.; et al. The serine/threonine kinase Pim-2 is a novel anti-apoptotic mediator in myeloma cells. Leukemia 2011, 25, 1182–1188.
- Brocke-Heidrich, K.; Kretzschmar, A.K.; Pfeifer, G.; Henze, C.; Löffler, D.; Koczan, D.; Thiesen, H.-J.; Burger, R.; Gramatzki, M.; Horn, F. Interleukin-6–dependent gene expression profiles in multiple myeloma INA-6 cells reveal a Bcl-2 family–independent survival pathway closely associated with Stat3 activation. Blood 2004, 103, 242–251.
- van Lohuizen, M.; Verbeek, S.; Krimpenfort, P.; Domen, J.; Saris, C.; Radaszkiewicz, T.; Berns, A. Predisposition to lymphomagenesis in pim-1 transgenic mice: Cooperation with c-myc and N-myc in murine leukemia virus-induced tumors. Cell 1989, 56, 673–682.
- Allen, J.D.; Verhoeven, E.; Domen, J.; Van Der Valk, M.; Berns, A. Pim-2 transgene induces lymphoid tumors, exhibiting potent synergy with c-myc. Oncogene 1997, 15, 1133–1141.
- Tian, Z.; Zhao, J.-J.; Tai, Y.-T.; Amin, S.B.; Hu, Y.; Berger, A.J.; Richardson, P.; Chauhan, D.; Anderson, K.C. Investigational agent MLN9708/2238 targets tumor-suppressor miR33b in MM cells. Blood 2012, 120, 3958–3967.
- Wu, L.; Xia, L.; Chen, X.; Ruan, M.; Li, L.; Xia, R. Long non-coding RNA LINC01003 suppresses the development of multiple myeloma by targeting miR-33a-5p/PIM1 axis. Leuk. Res. 2021, 106, 106565.
- Isaac, M.; Siu, A.; Jongstra, J. The oncogenic PIM kinase family regulates drug resistance through multiple mechanisms. Drug Resist. Updat. 2011, 14, 203–211.
- Xie, Y.; Xu, K.; Linn, D.E.; Yang, X.; Guo, Z.; Shimelis, H.; Nakanishi, T.; Ross, D.D.; Chen, H.; Fazli, L.; et al. The 44-kDa Pim-1 Kinase Phosphorylates BCRP/ABCG2 and Thereby Promotes Its Multimerization and Drug-resistant Activity in Human Prostate Cancer Cells. J. Biol. Chem. 2008, 283, 3349–3356.
- Hideshima, T.; Catley, L.; Yasui, H.; Ishitsuka, K.; Raje, N.; Mitsiades, C.; Podar, K.; Munshi, N.C.; Chauhan, D.; Richardson, P.G.; et al. Perifosine, an oral bioactive novel alkylphospholipid, inhibits Akt and induces in vitro and in vivo cytotoxicity in human multiple myeloma cells. Blood 2006, 107, 4053–4062.
- McMillin, D.W.; Ooi, M.; Delmore, J.; Negri, J.; Hayden, P.; Mitsiades, N.; Jakubikova, J.; Maira, S.-M.; Garcia-Echeverria, C.; Schlossman, R.; et al. Antimyeloma Activity of the Orally Bioavailable Dual Phosphatidylinositol 3-Kinase/Mammalian Target of Rapamycin Inhibitor NVP-BEZ235. Cancer Res. 2009, 69, 5835–5842.
- Hammerman, P.S.; Fox, C.J.; Birnbaum, M.; Thompson, C.B. Pim and Akt oncogenes are independent regulators of hematopoietic cell growth and survival. Blood 2005, 105, 4477–4483.
- Lu, J.; Zavorotinskaya, T.; Dai, Y.; Niu, X.-H.; Castillo, J.; Sim, J.; Yu, J.; Wang, Y.; Langowski, J.L.; Holash, J.; et al. Pim2 is required for maintaining multiple myeloma cell growth through modulating TSC2 phosphorylation. Blood 2013, 122, 1610–1620.
- Raab, M.S.; Thomas, S.K.; Ocio, E.M.; Guenther, A.; Goh, Y.-T.; Talpaz, M.; Hohmann, N.; Zhao, S.; Xiang, F.; Simon, C.; et al. The first-in-human study of the pan-PIM kinase inhibitor PIM447 in patients with relapsed and/or refractory multiple myeloma. Leukemia 2019, 33, 2924–2933.
- Paíno, T.; González-Méndez, L.; San-Segundo, L.; Corchete, L.A.; Hernández-García, S.; Díaz-Tejedor, A.; Algarín, E.M.; Mogollón, P.; Sánchez, L.A.C.; Gutiérrez, N.C.; et al. Protein Translation Inhibition is Involved in the Activity of the Pan-PIM Kinase Inhibitor PIM447 in Combination with Pomalidomide-Dexamethasone in Multiple Myeloma. Cancers 2020, 12, 2743.
- Koblish, H.; Li, Y.-L.; Shin, N.; Hall, L.; Wang, Q.; Wang, K.; Covington, M.; Marando, C.; Bowman, K.; Boer, J.; et al. Preclinical characterization of INCB053914, a novel pan-PIM kinase inhibitor, alone and in combination with anticancer agents, in models of hematologic malignancies. PLoS ONE 2018, 13, e0199108.
- Mihaly, S.R.; Ninomiya-Tsuji, J.; Morioka, S. TAK1 control of cell death. Cell Death Differ. 2014, 21, 1667–1676.
- Sakurai, H. Targeting of TAK1 in inflammatory disorders and cancer. Trends Pharmacol. Sci. 2012, 33, 522–530.
- Mukhopadhyay, H.; Lee, N.Y. Multifaceted roles of TAK1 signaling in cancer. Oncogene 2019, 39, 1402–1413.
- Tenshin, H.; Teramachi, J.; Oda, A.; Amachi, R.; Hiasa, M.; Bat-Erdene, A.; Watanabe, K.; Iwasa, M.; Harada, T.; Fujii, S.; et al. TAK1 inhibition subverts the osteoclastogenic action of TRAIL while potentiating its antimyeloma effects. Blood Adv. 2017, 1, 2124–2137.
- Salazar, L.; Kashiwada, T.; Krejci, P.; Meyer, A.N.; Casale, M.; Hallowell, M.; Wilcox, W.R.; Donoghue, D.J.; Thompson, L.M. Fibroblast Growth Factor Receptor 3 Interacts with and Activates TGFβ-Activated Kinase 1 Tyrosine Phosphorylation and NFκB Signaling in Multiple Myeloma and Bladder Cancer. PLoS ONE 2014, 9, e86470.

