 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Paloma Perez | + 1817 word(s) | 1817 | 2021-09-22 06:03:59 | | | |
| 2 | Nora Tang | + 575 word(s) | 2392 | 2021-10-27 05:23:00 | | |
Video Upload Options
Glucocorticoids (GCs) are steroid hormones that regulate the physiology of all mammalian tissues throughout life due to their diverse roles in development, growth, metabolism, and inflammation. In response to physiological cues and stressors, the hypothalamic-pituitary-adrenal (HPA) axis coordinates the systemic production and secretion of GCs from the adrenal glands in a circadian and stress-related manner to maintain tissue homeostasis. In turn, GCs mediate negative feed-back inhibiting secretion of the hypothalamic corticotropin-releasing hormone (CRH) and the adrenocorticotropic hormone (ACTH), limiting GC production.
1. Introduction
Glucocorticoids (GCs) are steroid hormones that regulate the physiology of all mammalian tissues throughout life due to their diverse roles in development, growth, metabolism, and inflammation [1][2][3][4]. In response to physiological cues and stressors, the hypothalamic-pituitary-adrenal (HPA) axis coordinates the systemic production and secretion of GCs from the adrenal glands in a circadian and stress-related manner to maintain tissue homeostasis. In turn, GCs mediate negative feed-back inhibiting secretion of the hypothalamic corticotropin-releasing hormone (CRH) and the adrenocorticotropic hormone (ACTH), limiting GC production.
The homeostatic control exerted by the HPA axis fits with an inverted U-shaped dose-response curve, where the equilibrium is achieved in the central range of the curve (optimal) while both GC excess or deficiency occur on either side of the curve (suboptimal effects) [2][5][6]. The disruption of this central HPA axis, due to pathophysiological triggers such as chronic stress, inflammation, or by prolonged exogenous GC treatments, results in abnormal endogenous GC levels. These GC imbalances contribute to disease and disease susceptibility in tissue-specific manners and with variable degrees of severity, by mechanisms that are not completely deciphered. Besides the adrenal GC production, certain tissues such as the thymus, intestine, brain, and skin, express functional equivalents of the HPA axis that allow for GC synthesis de novo [7][8]. This local GC production is increasingly recognized as a mechanism mediating rapid and critical control of immune activation [9].
Synthetic GCs are commonly prescribed to treat chronic inflammatory conditions including respiratory, autoimmune, and cutaneous diseases, as well as cancers of the hematopoietic lineage, mostly leukaemias [10]. The increasing number of GC-based prescriptions (around 3% of EU population uses GCs annually) [11] represents a high economic burden for health care systems, with demands rising as geriatric population and chronic diseases increase. However, despite their efficacy, long-term treatments and/or high doses with GCs trigger adverse effects, impacting metabolism (obesity, diabetes, and osteoporosis), and increasing susceptibility to stress and infections [10][12]. Another important limitation in the management of inflammatory diseases is resistance to GCs due to the lack of response or loss of efficacy over time [6][13][14][15]. The degrees of GC insensitivity are variable and highly dependent on the disease; for instance, while almost all patients with chronic obstructive pulmonary disease (COPD) and sepsis experience GC resistance, percentages are variable in patients with rheumatoid arthritis (30%), primary acute lymphoblastic leukaemia (ALL) (10–30%), or asthma (4–10%) [6].
Both endogenous and pharmacological GCs act through a dual system formed by the corticosteroid receptors GC receptor (GR/NR3C1) and mineralocorticoid receptor (MR/NR3C2), structurally and functionally close members of the nuclear receptor (NR) subclass NR3C that, upon hormone binding, act as ligand-activated transcription factors (TFs) [16][17][18][19][20]. While GR is ubiquitously expressed and almost exclusively activated by GCs, MR expression pattern is more restricted and can bind GCs and the mineralocorticoid aldosterone with similar high affinity. In tissues where GR and MR co-express, the selective activation of these TFs is achieved by pre-receptor mechanisms that modulate the local availability of active GCs. The enzymes 11β-hydroxysteroid dehydrogenase (HSD11B) type I and II, which catalyse the interconversion between active (cortisol) and inactive (cortisone) forms, represent a major regulatory mechanism for receptor selectivity [19][21][22].
2. Structure and Function of the Glucocorticoid Receptor (GR)
The oligomerization of GR is still a matter of intense debate [23][24]. It has been observed that several of the GR domains contribute to self-association critical for functionality. Both the DBD ( Figure 1 C) and LBD ( Figure 1 D–F) domains dimerize and their crystal structures have been solved [25]. Despite common acceptance of DBD dimerization, LBD oligomerization is still debated with several models based on experimental structural data using X-ray crystallography and complimentary in silico methods [26]. Additionally, recent data in living cells obtained using the number and brightness technique have shown that the oligomeric state of GR bound to DNA may exhibit a more complex scenario where a monomer to dimer to tetramer transition is likely to occur [27]. To date, no detailed structural information is available either by X-ray crystallography or electron microscopy (EM) for full-length GR. However, recent EM data reported for the related androgen and oestrogen receptors may allow us to speculate that GR full length structure may resemble that of AR considering the higher degree of conservation of residues [28]. Furthermore, EM data have recently been published depicting how chaperones recognize the LBD of GR [29].
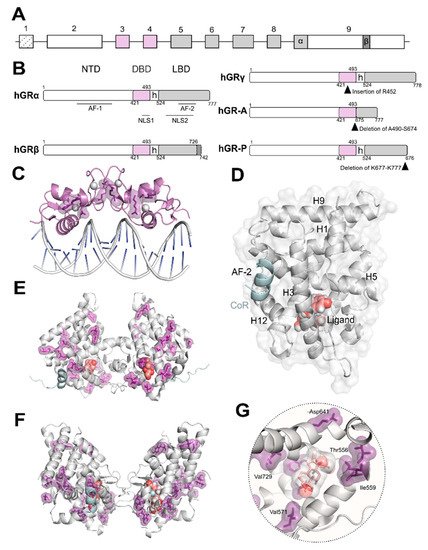
Upon ligand binding, GR dissociates from cytoplasmic heterocomplexes that include chaperones (HSP90, HSP70, and p23) and immunophilins (FKBP51 and FKBP52), undergoes PTMs, and translocates to the nucleus to regulate gene expression [4][10]. The GC-induced response is normally terminated by the autologous downregulation of GR following protein phosphorylation and subsequent ubiquitination through a PEST motif (rich in proline [P], glutamic acid [E], serine [S], and threonine [T]) in its C-terminus [4][10]. In a given cell type, the selective interactions of GR with TFs, co-regulators and/or chromatin modifying proteins determine receptor functionality [18][30][31][32]. This in part explains why different cell types vary in GR genomic binding and transcriptomic profile despite the ubiquitous expression of GR [33][34].
It is well established that GR anti-inflammatory properties rely on both DNA-binding–dependent and –independent receptor functions [33][35][36]. GR activates transcription of anti-inflammatory genes such as glucocorticoid induced leucine zipper ( GILZ/TSC22D3 ) by binding to GREs in regulatory regions, and represses expression of pro-inflammatory genes such as Il1-β by binding negative (n)GREs, preventing the assembly of an active transcription complex. GR binding has also been reported at inverted repeat (IR) nGREs, unrelated to classical GREs [36][37]. Also, the expression of inflammatory genes can be suppressed by the recruitment of histone deacetylases (HDAC) to acetylated GR complexes bound to GREs.
On the other hand, GR can regulate gene expression through protein–protein interactions (or tethering) to pro-inflammatory TFs (typically NF- κB or AP-1) bound to their respective DNA binding sites by a mechanism known as transrepression [4][35][36]. GR PTMs such as SUMOylation are required for GR-mediated inhibition of inflammatory genes via repression of IR nGRE genes as well as transrepression of NF-κB /AP-1-dependent transcription [10][18]. Besides their classic anti- inflammatory actions, GCs can also act as pro-inflammatory mediators, and even exert dual roles depending on the pathophysiological context [38]. Indeed, transcriptomic and cistromic studies have revealed that GR co-recruitment with key inflammatory TFs such as AP-1, NF-κB, or STATs, results in enhanced transcription at a subset of co-regulated targets [33]. Among the extensive crosstalk between GR and other signalling pathways, this review is focused on the mutual interference of GR with the mitogen-activated protein kinases (MAPKs).
3. Mutual Interference between GR and Mitogen-Activated Protein Kinases (MAPKs)
MAPKs are serine-threonine protein kinases that act as the meeting point for multiple upstream signalling pathways representing the final step of a phosphorylation cascade known as the MAPK module. In response to pleiotropic signals, including those from inflammatory cytokines, MAPKs dissociate from the MAPK module and can translocate to the nucleus to target TFs such as AP-1 and NF-κB or phosphorylate downstream protein kinases or other substrates to regulate gene expression [39][40] ( Figure 2 ). Therefore, these kinases are critical in regulating inflammation although they also play key roles in regulating cell growth, apoptosis, and differentiation. In mammals, the MAPK family comprises extracellular signal-regulated kinases (ERK), c-Jun N-terminal kinases (JNK), and p38 MAPKs [40].
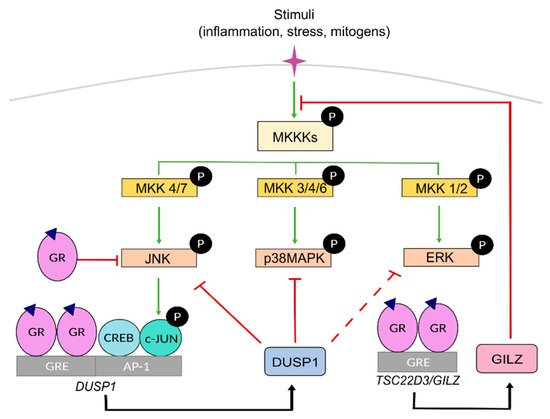
The ERK subfamily includes five members, among which ERK1 and ERK2 are the most extensively studied, being critical effectors of cell proliferation in response to growth factors. JNK family includes up to ten isoforms generated by alternative splicing from three genes ( Jnk 1-3). Importantly, JNK-mediated phosphorylation of the AP-1 component c-JUN is critical for AP-1-dependent transcription. The subgroup of p38 MAPK includes four isoforms (α, β, γ, δ), with different degrees of involvement in inflammatory disorders.
Within the MAPK module, MAPKs are activated by upstream specific kinases known as MAPK kinases, MKKs or MAP2Ks, through phosphorylation on tyrosine and threonine residues, which in turn are activated by MAPK kinase kinases, known as MKKKs or MAP3Ks, whose activation is triggered by several signals involving RAS GTPases. Specific combinations of scaffold proteins/MKKK/MKK/MAPK allow for specific cellular responses for diverse upstream events towards effector proteins. Each MKK isoform acts specifically onto downstream MAPKs. MKK1 and MKK2, MKK4 and MKK7, and MKK3, MKK4, and MKK6 activate ERK, JNK and p38 MAPK, respectively ( Figure 2 ).
MAPK activities can be inhibited by dual-specificity phosphatases (DUSPs or MAPK phosphatases MKP). Among them, DUSP1/MKP1 has a key role as a crucial anti-inflammatory mediator and gatekeeper of the immune response by dephosphorylating and targeting preferentially, but not exclusively, JNK and p38 MAPK [41][42]. However, under certain settings, the JNK/c-JUN pathway can also transcriptionally up-regulate DUSP1 to limit the expression of inflammatory genes, representing an important feedback regulatory loop [43][44][45][46][47] ( Figure 2 ).
4. GC Resistance due to the Crosstalk between GR and MAPK Signalling
In peripheral blood mononuclear cells (PBMCs) from severe asthmatic patients, GC-resistance correlates with higher levels of pro-inflammatory cytokines as well as increased expression and activity of the p38 α and β isoforms, relative to GC-responsive individuals [6][13][48]. The increased levels of cytokines in alveolar macrophages from asthmatic patients with reduced GC sensitivity result in the inhibition of GR function through its phosphorylation via p38α as well as the decreased induction of DUSP1 by GCs [13][49]. These findings are consistent with experimental models using macrophages from Dusp1 KO mice where the anti-inflammatory responses to GCs were reduced in vitro due to attenuated effects on gene expression and, as DUSP1 inactivates p38, as a consequence of increased p38 activation [50].
GCs also exert their anti-inflammatory effects by inhibiting the activity of phospholipases A2 (PLA2), which regulate the production of arachidonic acid, a precursor of lipid inflammatory mediators, playing important roles in many inflammatory diseases [51]. The reduced response to GCs in respiratory disorders such as acute lung injury may be due to reduced GR binding to cortisol and/or GRα overexpression, resulting in excessive activation of the MAPK cascade and insufficient regulation of downstream PLA2 pathway, therefore accentuating the pathologic response [52].
Many autoimmune diseases such as rheumatoid arthritis and inflammatory bowel diseases (IBD) show reduced effectiveness to routine treatments with GCs [53][54].
The mechanisms underlying GC resistance in IBDs include high levels of TNFα, IL-6, and IL-8, and low IL-10 levels, in steroid-resistant relative to sensitive patients, with activation of the MAPK/AP-1 and NF-κB pathways. MIF is also implicated in the pathogenesis of ulcerative colitis through activation of pro-inflammatory cytokines and subsequent anti-steroid effects [55][56]. As most cytokines are targets of major pro-inflammatory associated TFs, this scenario constitutes an auto-amplification loop for GC-resistance.
References
- Granner, D.K.; Wang, J.C.; Yamamoto, K.R. Regulatory Actions of Glucocorticoid Hormones: From Organisms to Mechanisms. Adv. Exp. Med. Biol. 2015, 872, 3–31.
- Cain, D.W.; Cidlowski, J.A. Specificity and Sensitivity of Glucocorticoid Signaling in Health and Disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 545–556.
- Whirledge, S.; DeFranco, D.B. Glucocorticoid Signaling in Health and Disease: Insights from Tissue-Specific GR Knockout Mice. Endocrinology 2018, 159, 46–61.
- Timmermans, S.; Souffriau, J.; Libert, C. A General Introduction to Glucocorticoid Biology. Front. Immunol. 2019, 10, 1–17.
- Nicolaides, N.C.; Charmandari, E. Novel Insights into the Molecular Mechanisms Underlying Generalized Glucocorticoid Resistance and Hypersensitivity Syndromes. Hormones 2017, 16, 124–138.
- Quax, R.A.; Manenschijn, L.; Koper, J.W.; Hazes, J.M.; Lamberts, S.W.J.; Van Rossum, E.F.C.; Feelders, R.A. Glucocorticoid Sensitivity in Health and Disease. Nat. Rev. Endocrinol. 2013, 9, 670–686.
- Talabér, G.; Jondal, M.; Okret, S. Extra-Adrenal Glucocorticoid Synthesis: Immune Regulation and Aspects on Local Organ Homeostasis. Mol. Cell. Endocrinol. 2013, 380, 89–98.
- Slominski, R.M.; Tuckey, R.C.; Manna, P.R.; Jetten, A.M.; Postlethwaite, A.; Raman, C.; Slominski, A.T. Extra-Adrenal Glucocorticoid Biosynthesis: Implications for Autoimmune and Inflammatory Disorders. Genes Immun. 2020, 21, 150–168.
- Merk, V.M.; Phan, T.S.; Brunner, T. Regulation of Tissue Immune Responses by Local Glucocorticoids at Epithelial Barriers and Their Impact on Interorgan Crosstalk. Front. Immunol. 2021, 12, 1–12.
- Vandewalle, J.; Luypaert, A.; De Bosscher, K.; Libert, C. Therapeutic Mechanisms of Glucocorticoids. Trends Endocrinol. Metab. 2018, 29, 42–54.
- Meijer, O.C.; Pereira, A.M. Three Percent Annually on Systemic Glucocorticoids: Facts, Worries and Perspectives. Eur. J. Endocrinol. 2019, 181, C23–C28.
- Vitellius, G.; Lombes, M. Genetics in Endocrinology: Glucocorticoid Resistance Syndrome. Eur. J. Endocrinol. 2020, 182, R15–R27.
- Barnes, P.J.; Adcock, I.M. Glucocorticoid Resistance in Inflammatory Diseases. Lancet 2009, 373, 1905–1917.
- Rodriguez, J.M.; Monsalves-Alvarez, M.; Henriquez, S.; Llanos, M.N.; Troncoso, R. Glucocorticoid Resistance in Chronic Diseases. Steroids 2016, 115, 182–192.
- Newton, R.; Shah, S.; Altonsy, M.O.; Gerber, A.N. Glucocorticoid and Cytokine Crosstalk: Feedback, Feedforward, and Co-Regulatory Interactions Determine Repression or Resistance. J. Biol. Chem. 2017, 292, 7163–7172.
- Kadmiel, M.; Cidlowski, J.A. Glucocorticoid Receptor Signaling in Health and Disease. Trends Pharmacol. Sci. 2013, 34, 518–530.
- Vandevyver, S.; Dejager, L.; Libert, C. Comprehensive Overview of the Structure and Regulation of the Glucocorticoid Receptor. Endocr. Rev. 2014, 35, 671–693.
- Weikum, E.R.; Knuesel, M.T.; Ortlund, E.A.; Yamamoto, K.R. Glucocorticoid Receptor Control of Transcription: Precision and Plasticity via Allostery. Nat. Rev. Mol. Cell Biol. 2017, 18, 159–174.
- Gomez-Sanchez, E.; Gomez-Sanchez, C.E. The Multifaceted Mineralocorticoid Receptor. Compr. Physiol. 2014, 4, 965–994.
- Jaisser, F.; Farman, N. Emerging Roles of the Mineralocorticoid Receptor in Pathology: Toward New Paradigms in Clinical Pharmacology. Pharmacol. Rev. 2016, 68, 49–75.
- Gathercole, L.L.; Lavery, G.G.; Morgan, S.A.; Cooper, M.S.; Sinclair, A.J.; Tomlinson, J.W.; Stewart, P.M. 11β-Hydroxysteroid Dehydrogenase 1: Translational and Therapeutic Aspects. Endocr. Rev. 2013, 34, 525–555.
- Chapman, K.; Holmes, M.; Seckl, J. 11β-Hydroxysteroid Dehydrogenases Intracellular Gate-Keepers of Tissue Glucocorticoid Action. Physiol. Rev. 2013, 93, 1139–1206.
- Yang, M.; Chen, J.; Wei, W. Dimerization of Glucocorticoid Receptors and Its Role in Inflammation and Immune Responses. Pharmacol. Res. 2021, 166, 105334.
- Presman, D.M.; Hager, G.L. More than Meets the Dimer: What Is the Quaternary Structure of the Glucocorticoid Receptor? Transcription 2017, 8, 32–39.
- Frank, F.; Okafor, C.D.; Ortlund, E.A. The First Crystal Structure of a DNA-Free Nuclear Receptor DNA Binding Domain Sheds Light on DNA-Driven Allostery in the Glucocorticoid Receptor. Sci. Rep. 2018, 8, 13497.
- Bianchetti, L.; Wassmer, B.; Defosset, A.; Smertina, A.; Tiberti, M.L.; Stote, R.H.; Dejaegere, A. Alternative Dimerization Interfaces in the Glucocorticoid Receptor-α Ligand Binding Domain. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1810–1825.
- Presman, D.M.; Ganguly, S.; Schiltz, R.L.; Johnson, T.A.; Karpova, T.S.; Hager, G.L. DNA Binding Triggers Tetramerization of the Glucocorticoid Receptor in Live Cells. Proc. Natl. Acad. Sci. USA 2016, 113, 8236–8241.
- Yu, X.; Yi, P.; Hamilton, R.A.; Shen, H.; Chen, M.; Foulds, C.E.; Mancini, M.A.; Ludtke, S.J.; Wang, Z.; O’Malley, B.W. Structural Insights of Transcriptionally Active, Full-Length Androgen Receptor Coactivator Complexes. Mol. Cell 2020, 79, 812–823.e4.
- Kirschke, E.; Goswami, D.; Southworth, D.; Griffin, P.R.; Agard, D.A. Glucocorticoid Receptor Function Regulated by Coordinated Action of the Hsp90 and Hsp70 Chaperone Cycles. Cell 2014, 157, 1685–1697.
- Ratman, D.; Vanden Berghe, W.; Dejager, L.; Libert, C.; Tavernier, J.; Beck, I.M.; De Bosscher, K. How Glucocorticoid Receptors Modulate the Activity of Other Transcription Factors: A Scope beyond Tethering. Mol. Cell. Endocrinol. 2013, 380, 41–54.
- Petta, I.; Dejager, L.; Ballegeer, M.; Lievens, S.; Tavernier, J.; De Bosscher, K.; Libert, C. The Interactome of the Glucocorticoid Receptor and Its Influence on the Actions of Glucocorticoids in Combatting Inflammatory and Infectious Diseases. Microbiol. Mol. Biol. Rev. 2016, 80, 495–522.
- Quatrini, L.; Ugolini, S. New Insights into the Cell- and Tissue-Specificity of Glucocorticoid Actions. Cell. Mol. Immunol. 2021, 18, 269–278.
- Sacta, M.A.; Chinenov, Y.; Rogatsky, I. Glucocorticoid Signaling: An Update from a Genomic Perspective. Annu. Rev. Physiol. 2016, 78, 155–180.
- Franco, L.M.; Gadkari, M.; Howe, K.N.; Sun, J.; Kardava, L.; Kumar, P.; Kumari, S.; Hu, Z.; Fraser, I.D.C.; Moir, S.; et al. Immune Regulation by Glucocorticoids Can Be Linked to Cell Type–Dependent Transcriptional Responses. J. Exp. Med. 2019, 216, 384–406.
- Desmet, S.J.; De Bosscher, K. Glucocorticoid Receptors: Finding the Middle Ground. J. Clin. Investig. 2017, 127, 1136–1145.
- Escoter-Torres, L.; Caratti, G.; Mechtidou, A.; Tuckermann, J.; Uhlenhaut, N.H.; Vettorazzi, S. Fighting the Fire: Mechanisms of Inflammatory Gene Regulation by the Glucocorticoid Receptor. Front. Immunol. 2019, 10, 1–17.
- Surjit, M.; Ganti, K.P.; Mukherji, A.; Ye, T.; Hua, G.; Metzger, D.; Li, M.; Chambon, P. Widespread Negative Response Elements Mediate Direct Repression by Agonist-Liganded Glucocorticoid Receptor. Cell 2011, 145, 224–241.
- Cain, D.W.; Cidlowski, J.A. Immune Regulation by Glucocorticoids. Nat. Rev. Immunol. 2017, 17, 233–247.
- Johnson, G.L.; Lapadat, R. Mitogen-Activated Protein Kinase Pathways Mediated by ERK, JNK, and P38 Protein Kinases. Science 2002, 298, 1911–1912.
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK Signal Transduction Pathways Activated by Stress and Inflammation: A 10-Year Update. Physiol. Rev. 2012, 92, 689–737.
- Lang, R.; Raffi, F.A.M. Dual-Specificity Phosphatases in Immunity and Infection: An Update. Int. J. Mol. Sci. 2019, 20, 2710.
- Hoppstädter, J.; Ammit, A.J. Role of Dual-Specificity Phosphatase 1 in Glucocorticoid-Driven Antiinflammatory Responses. Front. Immunol. 2019, 10, 1–8.
- Beck, I.M.E.; Vanden Berghe, W.; Vermeulen, L.; Yamamoto, K.R.; Haegeman, G.; De Bosscher, K. Crosstalk in Inflammation: The Interplay of Glucocorticoid Receptor-Based Mechanisms and Kinases and Phosphatases. Endocr. Rev. 2009, 30, 830–882.
- Sánchez-Tilló, E.; Comalada, M.; Xaus, J.; Farrera, C.; Valledor, A.F.; Caelles, C.; Lloberas, J.; Celada, A. JNK1 Is Required for the Induction of Mkp1 Expression in Macrophages during Proliferation and Lipopolysaccharide-Dependent Activation. J. Biol. Chem. 2007, 282, 12566–12573.
- Casals-Casas, C.; Álvarez, E.; Serra, M.; de la Torre, C.; Farrera, C.; Sánchez-Tilló, E.; Caelles, C.; Lloberas, J.; Celada, A. CREB and AP-1 Activation Regulates MKP-1 Induction by LPS or M-CSF and Their Kinetics Correlate with Macrophage Activation versus Proliferation. Eur. J. Immunol. 2009, 39, 1902–1913.
- Chi, H.; Barry, S.P.; Roth, R.J.; Wu, J.J.; Jones, E.A.; Bennett, A.M.; Flavell, R.A. Dynamic Regulation of Pro- and Anti-Inflammatory Cytokines by MAPK Phosphatase 1 (MKP-1) in Innate Immune Responses. Proc. Natl. Acad. Sci. USA 2006, 103, 2274–2279.
- Salojin, K.V.; Owusu, I.B.; Millerchip, K.A.; Potter, M.; Platt, K.A.; Oravecz, T. Essential Role of MAPK Phosphatase-1 in the Negative Control of Innate Immune Responses. J. Immunol. 2006, 176, 1899–1907.
- Kobayashi, Y.; Ito, K.; Kanda, A.; Tomoda, K.; Mercado, N.; Barnes, P.J. Impaired Dual-Specificity Protein Phosphatase DUSP4 Reduces Corticosteroid Sensitivity. Mol. Pharmacol. 2017, 91, 475–481.
- Khorasani, N.; Baker, J.; Johnson, M.; Chung, K.F.; Bhavsar, P.K. Reversal of Corticosteroid Insensitivity by P38 MAPK Inhibition in Peripheral Blood Mononuclear Cells from COPD. Int. J. COPD 2015, 10, 283–291.
- Abraham, S.M.; Lawrence, T.; Kleiman, A.; Warden, P.; Medghalchi, M.; Tuckermann, J.; Saklatvala, J.; Clark, A.R. Antiinflammatory Effects of Dexamethasone Are Partly Dependent on Induction of Dual Specificity Phosphatase 1. J. Exp. Med. 2006, 203, 1883–1889.
- Pniewska, E.; Pawliczak, R. The Involvement of Phospholipases A2 in Asthma and Chronic Obstructive Pulmonary Disease. Mediators Inflamm. 2013, 2013, 793505–793516.
- Kitsiouli, E.; Nakos, G.; Lekka, M.E. Phospholipase A2 Subclasses in Acute Respiratory Distress Syndrome. Biochim. Biophys. Acta 2009, 1792, 941–953.
- Straub, R.H. Rheumatoid Arthritis—A Neuroendocrine Immune Disorder: Glucocorticoid Resistance, Relative Glucocorticoid Defi Ciency, Low-Dose Glucocorticoid Therapy, and Insulin Resistance. Arthritis Res. Ther. 2014, 16 (Suppl. 2), I1.
- Dubois-Camacho, K.; Ottum, P.A.; Franco-Muñoz, D.; De La Fuente, M.; Torres-Riquelme, A.; Díaz-Jiménez, D.; Olivares-Morales, M.; Astudillo, G.; Quera, R.; Hermoso, M.A. Glucocorticosteroid Therapy in Inflammatory Bowel Diseases: From Clinical Practice to Molecular Biology. World J. Gastroenterol. 2017, 23, 6628–6638.
- de Iudicibus, S.; Franca, R.; Martelossi, S.; Ventura, A.; Decorti, G. Molecular Mechanism of Glucocorticoid Resistance in Inflammatory Bowel Disease. World J. Gastroenterol. 2011, 17, 1095–1108.
- Lorén, V.; Cabré, E.; Ojanguren, I.; Domènech, E.; Pedrosa, E.; García-Jaraquemada, A.; Mañosa, M.; Manyé, J. Interleukin-10 Enhances the Intestinal Epithelial Barrier in the Presence of Corticosteroids through P38 MAPK Activity in Caco-2 Monolayers: A Possible Mechanism for Steroid Responsiveness in Ulcerative Colitis. PLoS ONE 2015, 10, e0130921.

