 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Stephany El Hayek | + 2261 word(s) | 2261 | 2021-10-13 04:44:54 | | | |
| 2 | Jessie Wu | -3 word(s) | 2258 | 2021-10-26 06:20:21 | | |
Video Upload Options
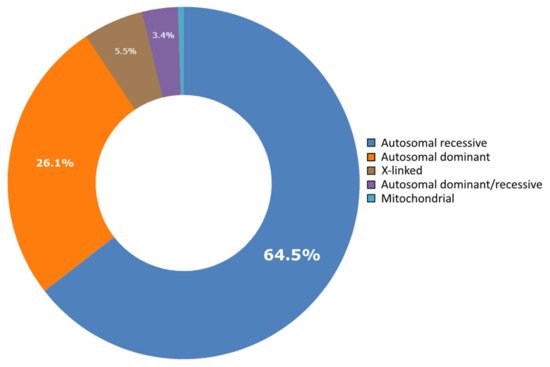
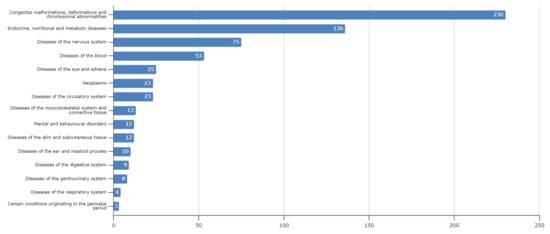
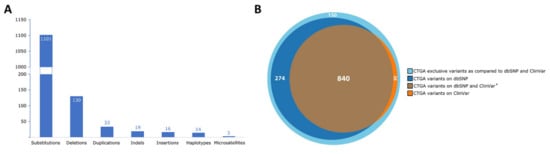
Lebanon has a high annual incidence of birth defects at 63 per 1000 live births, most of which are due to genetic factors. The Catalogue for Transmission Genetics in Arabs (CTGA) database, currently holds data on 642 genetic diseases and 676 related genes, described in Lebanese subjects. A subset of disorders (14/642) has exclusively been described in the Lebanese population, while 24 have only been reported in CTGA and not on OMIM. An analysis of all disorders highlights a preponderance of congenital malformations, deformations and chromosomal abnormalities and demonstrates that 65% of reported disorders follow an autosomal recessive inheritance pattern. In addition, our analysis reveals that at least 58 known genetic disorders were first mapped in Lebanese families. CTGA also hosts 1316 variant records described in Lebanese subjects, 150 of which were not reported on ClinVar or dbSNP. Most variants involved substitutions, followed by deletions, duplications, as well as in-del and insertion variants. This review of genetic data from the CTGA database highlights the need for screening programs, and is, to the best of our knowledge, the most comprehensive report on the status of genetic disorders in Lebanon to date.
1. Background
2. Current Insight on Lebanese Data on Genetic Disorders
References
- UNPFA. World Population Dashboard: Lebanon. Available online: https://www.unfpa.org/data/world-population/LB (accessed on 23 August 2021).
- UNHCR. 2020 Year-End Results. Available online: https://reporting.unhcr.org/lebanon (accessed on 23 August 2021).
- Tabar, P.; Denison, A. Diaspora Policies, Consular Services and Social Protection for Lebanese Citizens Abroad. In Migration and Social Protection in Europe and Beyond (Volume 3): A Focus on Non-EU Sending States; Lafleur, J.-M., Vintila, D., Eds.; Springer International Publishing: Cham, Germany, 2020; pp. 199–215.
- Christianson, A.; Howson, C.P.; Modell, B. March of Dimes: Global Report on Birth Defects; March of Dimes Birth Defects Foundation: White Plains, NY, USA, 2006.
- Al-Gazali, L.; Hamamy, H.; Al-Arrayad, S. Genetic disorders in the Arab world. BMJ 2006, 333, 831–834.
- Der Kaloustian, V.M. Genetic Disorders in Lebanon. In Genetic Disorders among Arab Populations; Teebi, A.S., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 377–441.
- Al-Gazali, L.; Hamamy, H. Consanguinity and dysmorphology in Arabs. Hum. Hered. 2014, 77, 93–107.
- Nakouzi, G.; Kreidieh, K.; Yazbek, S. A review of the diverse genetic disorders in the Lebanese population: Highlighting the urgency for community genetic services. J. Community Genet. 2015, 6, 83–105.
- Issam Khneisser, S.M.A.; Megarbane, A. Genetic Disorders in Lebanon: Challenges and Opportunities. In Genomics and Health in the Developing World; Kumar, D., Ed.; Oxford University Press: Oxford, UK, 2012.
- Der Kaloustian, V.M. Genetic diseases in Lebanon. Am. J. Med. Genet. 1986, 36, 65–68.
- CAGS. Catalogue for Transmission Genetics in Arabs Database. Available online: https://cags.org.ae/en/ctga-overview (accessed on 23 August 2021).
- Pipkin, A.C.; Pipkin, S.B. A pedigree of generalized lentigo. J. Hered. 1950, 41, 79–82.
- Bissar-Tadmouri, N.; Tadmouri, G.O. Bibliometric analyses of biomedical research outputs in Lebanon and the United Arab Emirates (1988–2007). Saudi Med. J. 2009, 30, 130–139.
- Megarbane, A.; Ghanem, I.; Romana, S.; Gosset, P.; Caillaud, C. Congenital contractures, short stature, abnormal face, microcephaly, scoliosis, hip dislocation, and severe psychomotor retardation in two unrelated girls. A new MCA/MR syndrome? Genet. Couns. 2002, 13, 123–131.
- Megarbane, A.; Haddad-Zebouni, S.; Nabbout, R.; Khoury, A.H.; Traboulsi, E.I. Microcephaly, colobomatous microphthalmia, short stature, and severe psychomotor retardation in two male cousins: A new MCA/MR syndrome? Am. J. Med. Genet. 1999, 83, 82–87.
- Wakim, V.; Nair, P.; Delague, V.; Bizzari, S.; Al-Ali, M.T.; Castro, C.; Alicia, G.; Stephany, E.-H.; André, M.M. SOX11-related syndrome: Report on a new case and review. Clin. Dysmorphol. 2021, 30, 44–49.
- Hana, S.; Karthik, D.; Shan, J.; El Hayek, S.; Chouchane, L.; Megarbane, A. A Report on a Family with TMTC3-Related Syndrome and Review. Case Rep. Med. 2020, 2020, 7163038.
- Bizzari, S.; El-Bazzal, L.; Nair, P.; Younan, A.; Stora, S.; Mehawej, C.; El-Hayek, S.; Delague, V.; Mégarbané, A. Recessive marfanoid syndrome with herniation associated with a homozygous mutation in Fibulin-3. Eur. J. Med. Genet. 2020, 63, 103869.
- Megarbane, A.; Rassi, S.; Estephan, F.; Kouba-Hreich, E. Post-natal short stature, short limbs, brachydactyly, facial abnormalities, and delayed bone age: A new syndrome? Am. J. Med. Genet. Part A 2004, 125A, 57–60.
- Oniya, O.; Neves, K.; Ahmed, B.; Konje, J.C. A review of the reproductive consequences of consanguinity. Eur. J. Obstet. Gynecol. Reprod. Biol. 2019, 232, 87–96.
- Nair, P.; Obaid, T.; Tadmouri, G.O. Genetic Disorders in Qatar: A CTGA Perspective. In Genetic Disorders in the Arab World: Qatar; Taleb Al Ali, M., Ed.; CAGS: Dubai, United Arab Emirates, 2012; Volume 4.
- Farah, R.A.; Nair, P.; Koueik, J.; Yammine, T.; Khalifeh, H.; Korban, R.; Agnes, C.; Claudia, K.; Catherine, D.-D.; Eliane, C.; et al. Clinical and Genetic Features of Patients with Fanconi Anemia in Lebanon and Report on Novel Mutations in the FANCA and FANCG Genes. J. Pediatr. Hematol./Oncol. 2021, 43, e727–e735.
- Farra, C.; Badra, R.; Fares, F.; Muwakkit, S.; Dbaibo, G.; Dabbous, I.; Ashkar, H.; Mounsef, C.; Abboud, M.R. Alpha thalassemia allelic frequency in Lebanon. Pediatr. Blood Cancer 2015, 62, 120–122.
- Inati, A.; Zeineh, N.; Isma’eel, H.; Koussa, S.; Gharzuddine, W.; Taher, A. Beta-thalassemia: The Lebanese experience. Clin. Lab. Haematol. 2006, 28, 217–227.
- Jelwan, Y.A.; Asbeutah, A.A.A.; Welty, F.K. Comprehensive Review of Cardiovascular Diseases, Diabetes, and Hypercholesterolemia in Lebanon. Cardiol. Rev. 2020, 28, 73–83.
- Maddirevula, S.; Shamseldin, H.E.; Sirr, A.; AlAbdi, L.; Lo, R.S.; Ewida, N.; Al-Qahtani, M.; Hashem, M.; Abdulwahab, F.; Aboyousef, O.; et al. Exploiting the Autozygome to Support Previously Published Mendelian Gene-Disease Associations: An Update. Front. Genet. 2020, 11, 580484.
- Romdhane, L.; Mezzi, N.; Hamdi, Y.; El-Kamah, G.; Barakat, A.; Abdelhak, S. Consanguinity and Inbreeding in Health and Disease in North African Populations. Annu. Rev. Genom. Hum. Genet. 2019, 20, 155–179.
- Alkuraya, F.S. Autozygome decoded. Genet. Med. 2010, 12, 765–771.
- Megarbane, A. Clinical genetics revisited: Effect of new techniques (next-generation sequencing, comparative genomic hybridization) on previous diagnoses. Middle East. J. Med. Genet. 2018, 7, 1–6.
- Lim, S.C.; Smith, K.R.; Stroud, D.; Compton, A.; Tucker, E.; Dasvarma, A.; Gandolfo, L.C.; Marum, J.E.; McKenzie, M.; Peters, H.L.; et al. A founder mutation in PET100 causes isolated complex IV deficiency in Lebanese individuals with Leigh syndrome. Am. J. Hum. Genet. 2014, 94, 209–222.
- Jalkh, N.; Corbani, S.; Haidar, Z.; Hamdan, N.; Farah, E.; Abou Ghoch, J.; Ghosn, R.; Salem, N.; Fawaz, A.; Khayat, C.D.; et al. The added value of WES reanalysis in the field of genetic diagnosis: Lessons learned from 200 exomes in the Lebanese population. BMC Med. Genom. 2019, 12, 11.
- Mansour, H.; Sabbagh, S.; Bizzari, S.; El-Hayek, S.; Chouery, E.; Gambarini, A.; Gencik, M.; Mégarbané, A. The Lebanese Allele in the PET100 Gene: Report on Two New Families with Cytochrome c Oxidase Deficiency. J. Pediatr. Genet. 2019, 8, 172–178.
- Riley, L.G.; Cowley, M.J.; Gayevskiy, V.; Minoche, A.E.; Puttick, C.; Thorburn, D.; Rius, R.; Compton, A.; Menezes, M.J.; Bhattacharya, K.; et al. The diagnostic utility of genome sequencing in a pediatric cohort with suspected mitochondrial disease. Genet. Med. 2020, 22, 1254–1261.
- Fahed, A.C.; Safa, R.M.; Haddad, F.F.; Bitar, F.F.; Andary, R.R.; Arabi, M.T.; Azar, S.T.; Nemer, G. Homozygous familial hypercholesterolemia in Lebanon: A genotype/phenotype correlation. Mol. Genet. Metab. 2011, 102, 181–188.
- Fahed, A.C.; Bitar, F.F.; Khalaf, R.I.; Moubarak, E.M.; Azar, S.T.; Nemer, G.M. The Lebanese allele at the LDLR in normocholesterolemic people merits reconsideration of genotype phenotype correlations in familial hypercholesterolemia. Endocrine 2012, 42, 445–448.
- Dos Santos, J.E.; Zago, M.A. Familial hypercholesterolemia in Brazil. Atheroscler. Suppl. 2003, 4, 1–2.
- Fahed, A.C.; Khalaf, R.; Salloum, R.; Andary, R.R.; Safa, R.; El-Rassy, I.; Moubarak, E.; Azar, S.T.; Bitar, F.F.; Nemer, G. Variable expressivity and co-occurrence of LDLR and LDLRAP1 mutations in familial hypercholesterolemia: Failure of the dominant and recessive dichotomy. Mol. Genet. Genom. Med. 2016, 4, 283–291.
- Fahed, A.C.; Shibbani, K.; Andary, R.R.; Arabi, M.T.; Habib, R.H.; Nguyen, D.D.; Haddad, F.F.; Moubarak, E.; Nemer, G.; Azar, S.T.; et al. Premature Valvular Heart Disease in Homozygous Familial Hypercholesterolemia. Cholesterol 2017, 2017, 3685265.
- Johnson, J.O.; Gibbs, J.R.; Megarbane, A.; Urtizberea, J.A.; Hernandez, D.G.; Foley, A.R.; Arepalli, S.; Pandraud, A.; Simón-Sánchez, J.; Clayton, P.; et al. Exome sequencing reveals riboflavin transporter mutations as a cause of motor neuron disease. Brain 2012, 135, 2875–2882.
- Srour, M.; Putorti, M.L.; Schwartzentruber, J.; Bolduc, V.; Shevell, M.I.; Poulin, C.; O’ferrall, E.; Buhas, D.; Majewski, J.; Brais, B. Mutations in riboflavin transporter present with severe sensory loss and deafness in childhood. Muscle Nerve 2014, 50, 775–779.
- Agarwal, A.K.; Simha, V.; Oral, E.A.; Moran, S.A.; Gorden, P.; O’Rahilly, S.; Zaidi, Z.; Gurakan, F.; Arslanian, S.A.; Klar, A.; et al. Phenotypic and genetic heterogeneity in congenital generalized lipodystrophy. J. Clin. Endocrinol. Metab. 2003, 88, 4840–4847.
- Van Maldergem, L.; Magre, J.; Khallouf, T.E.; Gedde-Dahl, T.; Delépine, M.; Trygstad, O.; Seemanova, E.; Stephenson, T.; Albott, C.S.; Bonnici, F.; et al. Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy. J. Med. Genet. 2002, 39, 722–733.
- Feng, J.Q.; Ward, L.M.; Liu, S.; Lu, Y.B.; Xie, Y.X.; Yuan, B.Z.; Yu, X.J.; Rauch, F.; Davis, S.I.; Zhang, S.B.; et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat. Genet. 2006, 38, 1310–1315.
- Gannage-Yared, M.H.; Makrythanasis, P.; Chouery, E.; Sobacchi, C.; Mehawej, C.; Santoni, F.A.; Guipponi, M.; Antonarakis, S.E.; Hamamy, H.; Mégarbané, A. Exome sequencing reveals a mutation in DMP1 in a family with familial sclerosing bone dysplasia. Bone 2014, 68, 142–145.
- Lorenz-Depiereux, B.; Bastepe, M.; Benet-Pages, A.; Amyere, M.; Wagenstaller, J.; Müller-Barth, U.; Badenhoop, K.; Kaiser, S.M.; Rittmaster, R.S.; Shlossberg, A.H.; et al. DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat. Genet. 2006, 38, 1248–1250.
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291.
- Abou Tayoun, A.N.; Rehm, H.L. Genetic variation in the Middle East-an opportunity to advance the human genetics field. Genome Med. 2020, 12, 116.
- Thauvin-Robinet, C.; Lee, J.S.; Lopez, E.; Herranz-Pérez, V.; Shida, T.; Franco, B.; Jego, L.; Ye, F.; Pasquier, L.; Loget, P.; et al. The oral-facial-digital syndrome gene C2CD3 encodes a positive regulator of centriole elongation. Nat. Genet. 2014, 46, 905–911.
- Alby, C.; Piquand, K.; Huber, C.; Megarbané, A.; Ichkou, A.; Legendre, M.; Pelluard, F.; Encha-Ravazi, F.; Abi-Tayeh, G.; Bessières, B.; et al. Mutations in KIAA0586 Cause Lethal Ciliopathies Ranging from a Hydrolethalus Phenotype to Short-Rib Polydactyly Syndrome. Am. J. Hum. Genet. 2015, 97, 311–318.
- Stoetzel, C.; Laurier, V.; Davis, E.E.; Muller, J.; Rix, S.; Badano, J.L.; Leitch, C.C.; Salem, N.; Chouery, E.; Corbani, S.; et al. BBS10 encodes a vertebrate-specific chaperonin-like protein and is a major BBS locus. Nat. Genet. 2006, 38, 521–524.
- Rutland, J.; de Iongh, R.U. Random ciliary orientation. A cause of respiratory tract disease. N. Eng. J. Med. 1990, 323, 1681–1684.
- Megarbane, A.; Ghanem, I.; Waked, N.; Dagher, F. A newly recognized autosomal recessive syndrome with short stature and oculo-skeletal involvement. Am. J. Med. Genet. Part A 2006, 140, 1491–1496.

