 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Karthika Rajeeve | + 1231 word(s) | 1231 | 2021-10-19 04:37:59 | | | |
| 2 | Jason Zhu | -166 word(s) | 1065 | 2021-10-21 03:42:27 | | | | |
| 3 | Jason Zhu | -166 word(s) | 1065 | 2021-10-21 03:47:49 | | | | |
| 4 | Jason Zhu | Meta information modification | 1065 | 2021-10-25 03:05:43 | | | | |
| 5 | Jason Zhu | + 3 word(s) | 1068 | 2021-10-26 11:48:37 | | |
Video Upload Options
Epidemiological evidence reveal a very close association of malignancies with chronic inflammation as a result of persistent bacterial infection. Recently, more studies have provided experimental evidence for an etiological role of bacterial factors disposing infected tissue towards carcinoma. When healthy cells accumulate genomic insults resulting in DNA damage, they may sustain proliferative signalling, resist apoptotic signals, evade growth suppressors, enable replicative immortality, and induce angiogenesis, thus boosting active invasion and metastasis.
1. Introduction
As a result of both intra and intergenomic diversification, H. pylori strains have shown to appear with a broad genetic diversity [1]. Certain strain-specific proteins such as vacuolating cytotoxin A (VacA) and cytotoxin-associated gene A (CagA) are shown to have increased risk towards carcinoma [2][3][4][5]. VacA is a channel forming toxin capable of inducing vacuoles into epithelial cells and has long been associated with the development of gastric inflammation [2]. While the exact oncogenic mechanisms have not yet been established, potential pathways have been suggested: VacA was found to cause apoptosis in gastro-epithelial cells leading to increased cell proliferation or a possible entry point for other carcinogens into the gastric mucosa [2]. VacA can activate the phosphoinositide 3-kinase (PI3K/AKT) pathway, resulting in the inhibition of glycogen synthase kinase 3 beta (GSK3β) via phosphorylation, with the subsequent dissociation of the GSK3β/β-catenin complex [6].
CagA activates the extracellular signal-regulated kinases/mitogen-activated protein kinase (ERK/MAPK) pathway through SHP2, resulting in morphological changes in the epithelium called hummingbird phenotype in a Ras-independent manner [7]. CagA also interacts with the E-cadherin/β-catenin complex resulting in nuclear accumulation of β-catenin leading to transdifferentiation of gastric epithelial cells [8]. The bacteria can induce the epithelial-to-mesenchymal transition (EMT) mediated by Snail, a transcriptional repressor of E-Cadherin expression, in gastric epithelial cells by GSK3β depletion [9]. The tumour suppressor TP53 undergoes proteasomal degradation leading to DNA damage in the presence of CagA [10]. Most interestingly, the transgenic expression of CagA in mice induces gastrointestinal and hematopoietic neoplasms, revealing the direct carcinogenic potential of the protein [5].
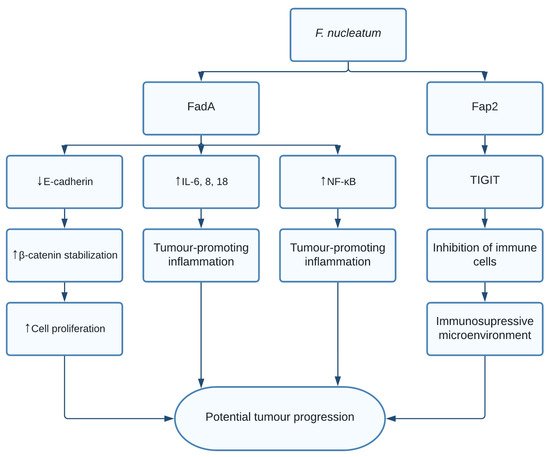
The role of F. nucleatum in CRC is thought to be through multiple different pathways. Fusobacterium adhesin A (FadA) was found to bind and inhibit E-cadherin in xenograft mice. E-cadherin is a tumour suppressor, causing activation of β-catenin signalling, previously mentioned to promote cell proliferation [11]. Additionally, FadA increased expression of nuclear factor-κB (NF-κB) and pro-inflammatory interleukins such as IL-6, IL-8, and IL-18 leading to a proinflammatory microenvironment [11]. Finally, F. nucleatum was shown to induce anticancer immune response evasion through fusobacterial apoptosis protein 2 (Fap2) [12]. Fap2 was found to bind the TIGIT immune receptor, a receptor expressed on all natural killer cells (NK cells) and other immune cells, thus inhibiting NK cell cytotoxicity [12]. Additionally, TIGIT was expressed on tumour-infiltrating lymphocytes and Fap2 was found to inhibit T cell activities, effectively protecting F. nucleatum from immune cell attacks ( Figure 1 ). Interestingly, it was shown that exosomes harbouring miR1246/92b-3p/27a-3p CXCL16 derived from F. nucleatum -infected colorectal cancer cells facilitated tumour metastasis [13]. Moreover, in vitro and in vivo studies show that the bacterial infection leads to upregulation of cytochrome p450 monooxygenase, which increases the invasiveness and migratory ability (EMT) of colorectal cancer cells via the TLR4/Keap1/NRF2 axis [14].
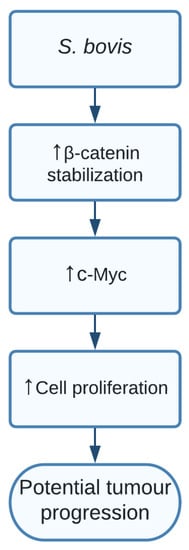
Current studies showed a clear indication that SG increased levels of β-catenin, c-Myc, and proliferative cell nuclear antigen (PCNA) resulting in cell proliferation in certain responsive cell lines. Additionally, silencing β-catenin through knockdown cells resulted in a completely abolished effect of SG on the increased cell proliferation, thereby indicating the important role of SG in activating β-catenin and promoting cell proliferation, resulting in potential tumour development. The pathway of how SG upregulated β-catenin is still unclear and should be further investigated [15] ( Figure 2 ).
2. Current Studies of Bacteria
Although a rather large group of bacteria has been shown to be associated with different cancer types, only H. pylori is currently accepted as a group 1 carcinogen, reflecting how the bacterial cancer field has not received the same amount of attention as the viral field. While some bacteria such as E. coli , S. bovis, and C. trachomatis have long been associated with cancer, studies focused on bacterial factors that directly predispose infected tissue towards carcinoma have only recently been reported. The bacterial effectors that can inflict mutations and result in malignancies in the infected tissue are yet to be discovered.
Several bacteria mentioned are commonly found in the human microbiome, yet they are associated with cancer. If a bacterium is causal for cancer, why is the cancer not seen more frequently? Even though cancer is one of the well-funded and focused research areas, much is still to be discovered. For a cell to turn malignant due to chronic infectious disease, the cell must accumulate the respective mutations in the DNA, or these epigenetic changes might imprint memory to activate indefinite survival signals leading to cell proliferation. Most of the showcased bacteria are capable of inflicting one or more of these hallmarks to an otherwise normal cell, but unless all the hallmarks are acquired, i.e., “the perfect storm has hit”, the cell may not be a fully functional cancer cell. If the bacteria truly are linked to the cause of cancer, they are not likely the only causal factor but instead a contribution in the multistep process required to fulfil the hallmarks of cancer, leaving the rest of the hallmarks to be acquired by other causes, such as radiation, smoking, aging, or inheritance Thus these chronic infections can increase the fitness of cells towards carcinoma.
Since many studies show that bacteria could be located at the site of tumour development, it is also debatable whether the bacteria might colocalize in the transformed tissue due to the excess nutrient availability and increased vascularization. This affinity for tumour tissue is exploited by scientists, by using attenuated strains of bacteria as adjuvants in the treatment of various neoplasms. The bacterial tropism for tumour microenvironment could also activate the innate and adaptive immune response of the host to clear malignant cells.
References
- Dorer, M.S.; Talarico, S.; Salama, N.R. Helicobacter pylori’s Unconventional Role in Health and Disease. PLoS Pathog. 2009, 5, e1000544.
- McClain, M.S.; Beckett, A.C.; Cover, T.L. Helicobacter pylori Vacuolating Toxin and Gastric Cancer. Toxins 2017, 9, 316.
- Polk, D.B.; Peek, R.M., Jr. Helicobacter pylori: Gastric cancer and beyond. Nat. Rev. Cancer 2010, 10, 403–414.
- Schulz, C.; Schütte, K.; Mayerle, J.; Malfertheiner, P. The role of the gastric bacterial microbiome in gastric cancer: Helicobacter pylori and beyond. Ther. Adv. Gastroenterol. 2019, 12, 1756284819894062.
- Ohnishi, N.; Yuasa, H.; Tanaka, S.; Sawa, H.; Miura, M.; Matsui, A.; Higashi, H.; Musashi, M.; Iwabuchi, K.; Suzuki, M.; et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc. Natl. Acad. Sci. USA 2008, 105, 1003–1008.
- Nakayama, M.; Hisatsune, J.; Yamasaki, E.; Isomoto, H.; Kurazono, H.; Hatakeyama, M.; Azuma, T.; Yamaoka, Y.; Yahiro, K.; Moss, J.; et al. Helicobacter pylori VacA-induced Inhibition of GSK3 through the PI3K/Akt Signaling Pathway. J. Biol. Chem. 2009, 284, 1612–1619.
- Higashi, H.; Nakaya, A.; Tsutsumi, R.; Yokoyama, K.; Fujii, Y.; Ishikawa, S.; Higuchi, M.; Takahashi, A.; Kurashima, Y.; Teishikata, Y.; et al. Helicobacter pylori CagA Induces Ras-independent Morphogenetic Response through SHP-2 Recruitment and Activation. J. Biol. Chem. 2004, 279, 17205–17216.
- Muratakamiya, N.; Kurashima, Y.; Teishikata, Y.; Yamahashi, Y.; Saito, Y.; Higashi, H.; Aburatani, H.; Akiyama, T.; Peek, R.M.; Azuma, T.; et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the β-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene 2007, 26, 4617–4626.
- Lee, D.-G.; Kim, H.S.; Lee, Y.S.; Kim, S.; Cha, S.; Ota, I.; Kim, N.H.; Cha, Y.H.; Yang, D.H.; Lee, Y.; et al. Helicobacter pylori CagA promotes Snail-mediated epithelial–mesenchymal transition by reducing GSK-3 activity. Nat. Commun. 2014, 5, 4423.
- Buti, L.; Spooner, E.; Van der Veen, A.G.; Rappuoli, R.; Covacci, A.; Ploegh, H.L. Helicobacter pylori cytotoxin-associated gene A (CagA) subverts the apoptosis-stimulating protein of p53 (ASPP2) tumor suppressor pathway of the host. Proc. Natl. Acad. Sci. USA 2011, 108, 9238–9243.
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum Promotes Colorectal Carcinogenesis by Modulating E-Cadherin/β-Catenin Signaling via its FadA Adhesin. Cell Host Microbe 2013, 14, 195–206.
- Gur, C.; Ibrahim, Y.; Isaacson, B.; Yamin, R.; Abed, J.; Gamliel, M.; Enk, J.; Bar-On, Y.; Stanietsky-Kaynan, N.; Coppenhagen-Glazer, S.; et al. Binding of the Fap2 Protein of Fusobacterium nucleatum to Human Inhibitory Receptor TIGIT Protects Tumors from Immune Cell Attack. Immunity 2015, 42, 344–355.
- Guo, S.; Chen, J.; Chen, F.; Zeng, Q.; Liu, W.-L.; Zhang, G. Exosomes derived from Fusobacterium nucleatum-infected colorectal cancer cells facilitate tumour metastasis by selectively carrying miR-1246/92b-3p/27a-3p and CXCL. Gut 2021, 70, 1507–1519.
- Kong, C.; Yan, X.; Zhu, Y.; Zhu, H.; Luo, Y.; Liu, P.; Ferrandon, S.; Kalady, M.F.; Gao, R.; He, J.; et al. Fusobacterium Nucleatum Promotes the Development of Colorectal Cancer by Activating a Cytochrome P450/Epoxyoctadecenoic Acid Axis via TLR4/Keap1/NRF2 Signaling. Cancer Res. 2021, 81, 4485–4498.
- Kumar, R.; Herold, J.L.; Schady, D.; Davis, J.; Kopetz, S.; Martinez-Moczygemba, M.; Murray, B.E.; Han, F.; Li, Y.; Callaway, E.; et al. Streptococcus gallolyticus subsp. gallolyticus promotes colorectal tumor development. PLoS Pathog. 2017, 13, e1006440.

